
二烯烃选择加氢非贵金属Ni-Mo-W催化剂
2014-09-22 08:31:30刘铁峰
东北石油大学学报 2014年2期
刘铁峰,郑 卓
(中国科学院 大连化学物理研究所,辽宁 大连 116023)
0 引言
二烯烃广泛存在于催化裂化汽油、热裂解汽油及一些富含烯烃的原料中.二烯烃性质非常活泼,除了自身容易聚合外,还同其他的烯烃发生反应,形成胶质及结焦前体;在汽油进一步加氢精制过程中,还导致催化剂床层堵塞、反应器压降上升和生产周期缩短等[1-2].因而,在对汽油及富含烯烃的原料进行加工前,必须将其中的二烯烃脱除,以避免二烯烃在催化剂上结焦而影响催化剂的使用寿命.国内外开发不同的选择加氢工艺,有选择性地脱除原料组分中的二烯烃,由于单烯烃也很容易加氢饱和[3],所以必须选择只对二烯烃加氢而对单烯烃不受影响的高选择性催化剂.
在选择加氢脱除二烯烃工艺的催化剂研究方面,主要有传统的Co-Mo或Ni-Mo催化剂[4],催化剂成本低,但是在反应活性和选择性方面不如贵金属催化剂的[5-8].贵金属催化剂以Pd系催化剂为主,Pd的活性和选择性优于Pt和Ni的[9],但是贵金属催化剂易中毒,并且成本较高.近年来,纳米态催化剂和非晶态合金催化剂越来越受到重视,Gilles Berhault等[10]研究负载于α-Al2O3上的纳米Pd催化剂,Vignea F[11]及Jugnet Y[12]等研究的Pt/Sn非晶态合金催化剂,在催化二烯选择加氢反应时具有较高的催化活性及选择性,但是无法克服易中毒的缺点.分子筛催化剂[13]成本低、不中毒,但是反应温度和压力较高.负载型非贵金属镍系催化剂[14-15]是目前应用比较广泛的一类催化剂,法国石油研究院(IFP)开发的LD-241催化剂[16]、中国石油化工股份有限公司北京燕山分公司石油化工研究院开发的BY-3催化剂[17],以及中国石油兰州化工研究中心开发的LY-2008催化剂[18]等,具有较好的加氢活性和选择性,但是反应温度和操作压力较高.非负载型镍系催化剂相关报导较少,笔者开发一种具有高活性的、高选择性的、反应温度和压力相对低的、相对于贵金属廉价的非负载型二烯烃选择加氢催化剂.
1 实验部分
1.1 催化剂制备
分别称取0.100mol硝酸镍、0.100mol硝酸锌和0.050mol硝酸铝,将它们溶于200mL水中形成水溶液,向其中缓慢滴加K+浓度为0.20mol/L的氢氧化钾与碳酸钾(0.10mol/L的氢氧化钾和0.05mol/L的碳酸钾)的混合溶液,调节pH=12,加热至反应温度80℃,生成混合反应液,回流反应25h;将反应得到的绿色沉淀过滤,得到NiZnAlK-LDH催化剂前体,将催化剂前体加入到200mL水中,配成前体浆液备用.
分别称取0.010mol钼酸铵和0.010mol偏钨酸铵,将它们溶于350mL水中,形成水溶液,将溶液加热至温度50~60℃,机械搅拌至体系呈无色透明溶液;然后量取含Ni2+(0.030mol)、Zn2+(0.030mol)、Al3+(0.015mol)自制的浆液前体,置于三口瓶中加热至温度80℃;将浆液加入到无色透明溶液中,形成混合反应液,在80~110℃温度回流反应1h;将反应得到的黄绿色沉淀过滤,在120℃温度烘干12h,在420℃温度焙烧4h,得到NiZnAlKMoW颗粒状棕黑色催化剂(Cat-A).
将Cat-A在研钵中磨碎,经打片机打片后,筛取20~40目催化剂颗粒置于干燥器中备用.
1.2 催化剂表征
采用Rigaku D/max-2500PC型X线衍射仪测定样品的XRD谱图,使用CuK射线源(1.540 6Å),广角XRD(5°~80°),管电压为40kV,管电流为100mA,扫描速率为5°/min.在MDI Jade5.0软件上进行谱图的定性分析,数据库使用PDF 2002.采用FEI Quanta 200F型扫描电子显微镜和Tecnai G2Spirit型透射电子显微镜,测定晶体形貌和晶粒尺寸.
1.3 催化剂性能评价
催化剂活性评价在10mL高压固定床反应装置上进行,采用内径为10mm的不锈钢管式反应器.以模型油为原料,考察Cat-A催化剂的反应性能.模型油以异戊二烯为模型化合物,将适量的异戊二烯溶于含有环己烯(质量分数为30%)的甲苯溶液中,通过马来酸酐法检测模型油双烯值等于1.0,采用安捷伦公司的GC6890型气相色谱仪分析烯烃(质量分数为30%).
1.4 二烯值检测方法
采用马来酸酐法测定模型油中的二烯值.在样品中加入过量的顺丁烯二酸酐甲苯溶液,加热回流反应3h,未反应的顺丁烯二酸酐水解后再用水抽提,用氢氧化钠标准溶液滴定未反应的酸,计算反应消耗的顺丁烯二酸酐的量,用碘值表示的值为双烯值.
2 结果与讨论
2.1 催化剂表征分析
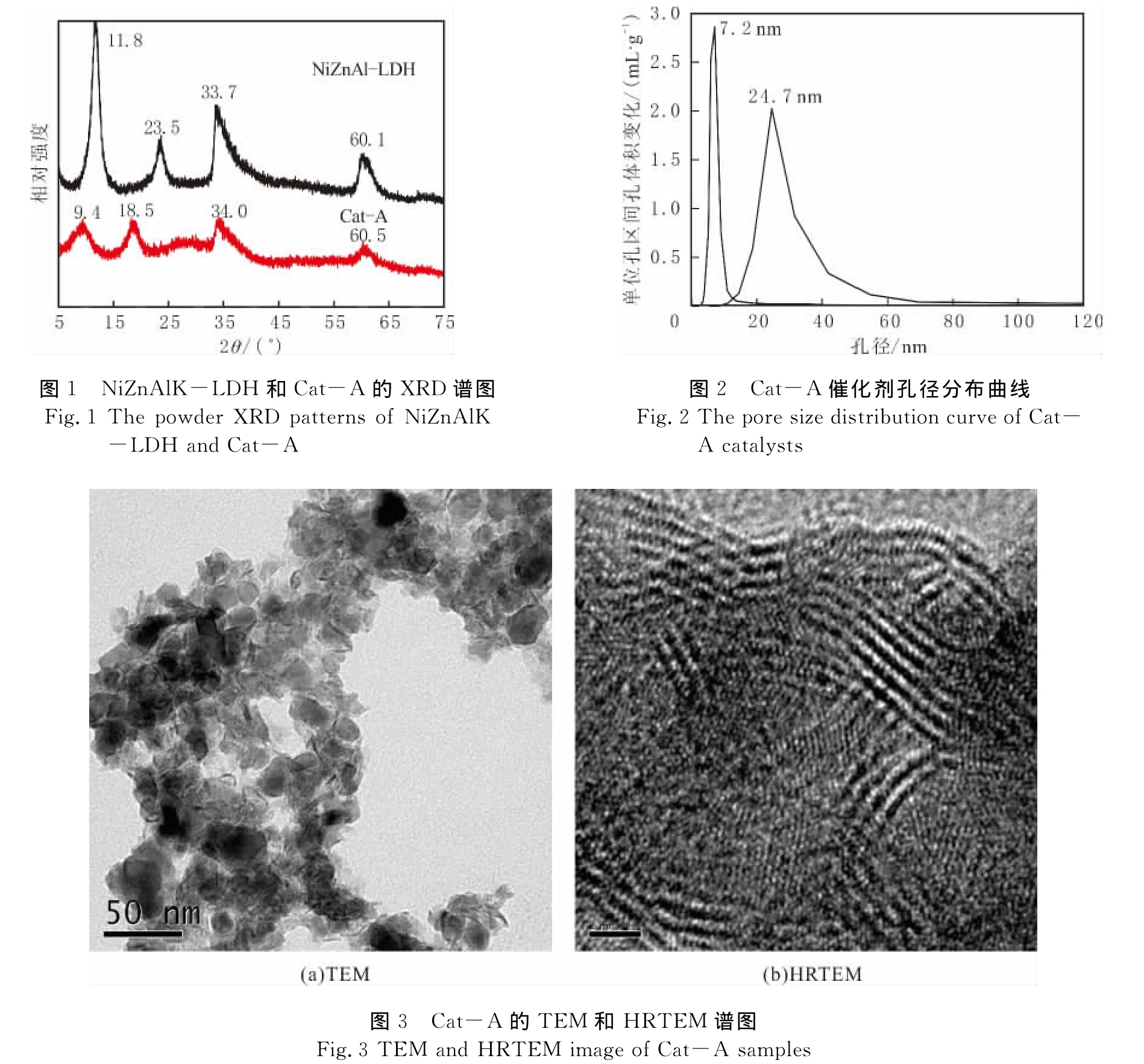
对制备的NiZnAlK-LDH催化剂前体和最终的Cat-A催化剂进行XRD表征,结果见图1.前体和催化剂在2θ=34°和60°附近处有明显的乱层堆叠结构特征衍射峰,证明合成具有乱层堆叠结构的催化剂前体及催化剂.在NiZnAlK-LDH催化剂前体的谱图中,出现4个比较强的衍射峰,分别位于2θ=11.8°、23.5°、33.7°、60.1°.通过含有 Mo、W 的阴离子交换后,如果 Mo、W 的阴离子只是简单地吸附在前体表面,则前体特征衍射峰不发生变化;在Cat-A催化剂中,位于2θ=9.4°、18.5°、34.0°、60.5°出现4个较宽的衍射峰,催化剂前体位于2θ=11.8°、23.5°的2个衍射峰向小角度位移,表明含Mo、W的阴离子已成功交换到NiZnAlK-LDH催化剂前体的层间,并且催化剂前体层结构得到很好保持.
通过Cat-A样品的氮吸附测定,可以获得催化剂的孔径分布,结果见图2.由图2可以看出,该催化剂具有双重孔分布模式,最可几孔径分别位于7.2nm和24.7nm.透射电镜(TEM)和高分辨透射电镜(HRTEM)表征进一步确认Cat-A的形貌特征,结果见图3.由图3(a)可以看出,该催化剂呈片状堆叠态,纳米粒子大小有很好的均一度;由图3(b)可以看出,硫化物催化剂的片晶长度在2~6nm之间,堆叠层数在4~8层之间,并且片晶中含有大量的中间断开的缺陷位,是催化剂具有高活性的重要原因.
在还原过程中,为了了解催化剂金属氧化物之间或金属氧化物与载体之间相互作用的信息,对催化剂Cat-A样品进行程序升温还原(TPR)表征,结果见图4.由图4可以看出,只有480℃温度处出现一个明显的耗氢峰,是Ni-Zn-Al-K-Mo-W固溶体氧化物的还原峰,证明该催化剂形成均一的NiMo(W)O4氧化物复合结构.这种复合结构在硫化后形成NiMo(W)S活性相,活性相在其他的文献中已有报道[19],显示超高的加氢脱硫活性,但选择加氢性能未见报道.还原反应从350℃温度开始,在达到480℃温度时耗氢量开始逐渐下降,还原反应结束.
2.2 催化剂反应性能评价
2.2.1 催化剂预硫化
将准备的10mL Cat-A催化剂装在10mL固定床反应器中.在进料前,先对催化剂进行固定反应器内预硫化,使用含质量分数为10%硫化氢的氢气对催化剂进行预硫化,硫化条件是氢分压为1.2MPa,氢油体积比为600,由室温程序升温至480℃恒温6h,硫化时间为16h.硫化完成后进油进行加氢脱二烯烃评价反应.
2.2.2 催化剂性能评价
在固定床反应装置上,考察不同氢油比、反应压力(p)、反应温度(t)及液时空速条件下催化剂Cat-A的选择加氢性能,通入模型油进行反应,稳定48h后取样分析,结果见表1.

由表1可以看出,在反应温度为100~130℃,氢分压为0.5~2.0MPa,氢油体积比为10~90NL/L,液时空速为1.0~4.0h-1操作条件下,催化剂能够有效地将模型油中的二烯值由1.0降至0.2以下.在高温为160℃,高氢分压为2.0MPa,高氢油体积比为90NL/L,低液时空速为1.0h-1时,原料中的单烯烃过度饱和,与模型油比较质量分数降低超过3%,不利于实际应用.
在优化选择加氢脱二烯烃反应操作后,在反应温度为130℃,氢分压为1.0MPa,氢油体积比为20 NL/L,液时空速为2.0h-1操作条件下,考察催化剂Cat-A使用寿命,装置运行250h后停止进料,部分分析结果见表2.
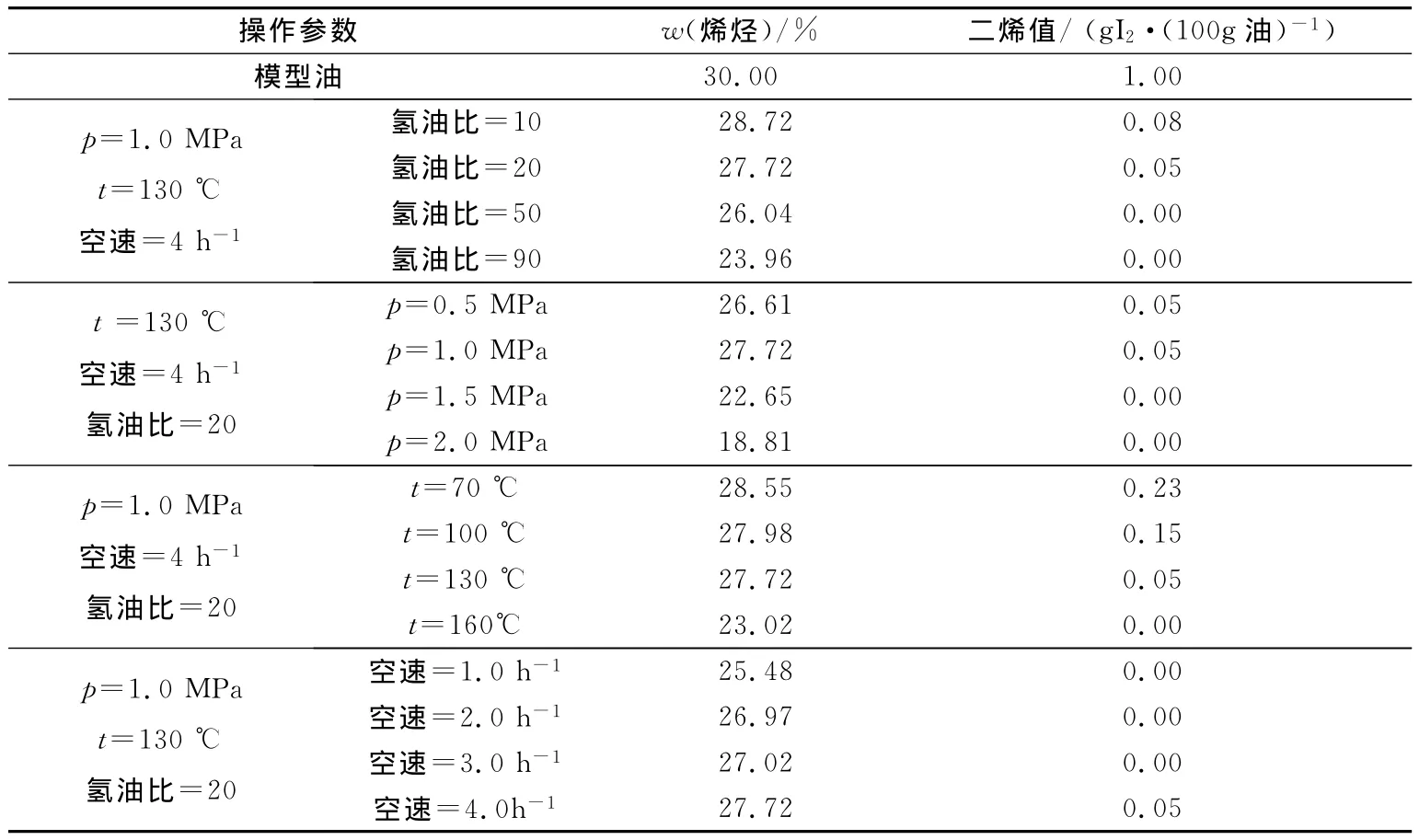
表1 模型油加氢实验结果Table 1 Results of hydrogenation for model oil after reaction

表2 反应后模型油二烯值Table 2 The diene value of model oil after reaction
2.3 结果分析
合成具有与传统催化剂不同物相结构的层状堆叠Cat-A催化剂,在硫化后形成高活性的NiMo(W)S活性相,并且在硫化物催化剂中存在大量的缺陷位,也是催化剂显示高的选择加氢活性的重要原因.Cat-A催化剂对模型油品中的二烯烃表现出好的加氢活性和选择性.目前,普遍采用的FO-35T系列脱二烯烃催化剂的平稳运行条件:氢油体积比为50NL/L,反应压力为2.05MPa,反应温度为200~210℃,液时空速为3.0h-1;Cat-A催化剂的平稳运行条件:氢油体积比为20NL/L,反应压力为1.0MPa,反应温度为100~130℃,液时空速为2.0h-1,在连续运转250h后仍保持优良的选择加氢性能.
3 结论
(1)以NiZnAlK-LDH为催化剂前驱体合成的Cat-A催化剂具有较好的加氢活性与选择性.
(2)Cat-A催化剂具有与传统催化剂不同的物相结构,形成NiMo(W)O4复合物结构,表征显示均一的层状堆叠结构.
(3)Cat-A催化剂在反应温度为130℃,反应压力为1.0MPa,氢油体积比为20NL/L,液时空速为2.0h-1反应条件下,能将二烯值为1.0的模型油脱二烯至0.2以下,单烯烃饱和质量分数下降在3%以下,并且能长期稳定运行.
(References):
[1]李大东.加氢处理工艺与工程[M].北京:中国石化出版社,2004:317-318.Li Dadong.Processes &engineering of residue hydrogenation[M].Beijing:China Petrochemical Press,2004:317-318.
[2]金谊,刘铁斌,魏民,等.催化裂化轻汽油在 Ni-K/Al2O3催化剂上选择加氢的研究[J].石油炼制与化工,2004,35(4):9-12.Jin Yi,Liu Tiebin,Wei Min,et al.Selective hydrogenation of FCC light naphtha on Ni-K/Al2O3catalyst[J].Petroleum Processing and Petrochemicals,2004,35(4):9-12.
[3]郑净植,奚强.炔烃及二烯烃选择加氢催化剂的研究进展[J].湖北化工,2003,1(1):4-5.Zheng Jingzhi,Xi Qiang.Advances in the application of catalysts to the selective hydrogenation of alkynes and dienes[J].Hubei Chemical Industry,2003,1(1):4-5.
[4]李建伟,李英霞,陈标华,等.Co-Mo/Al2O3催化剂上裂解汽油中单烯烃加氢宏观动力学[J].燃料化学学报,2006,34(2):170-174.Li Jianwei,Li Yingxia,Chen Biaohua,et al.Macrokinetics of olefin hydrogenation in pyrolysis gasoline over Co-Mo/Al2O3catalyst[J].Journal of Fuel Chemistry and Technology,2006,34(2):170-174.
[5]南军,柴永明,李彦鹏,等.Pd/Al2O3催化剂用于连续重整汽油全馏分加氢的失活分析[J].工业催化,2007,15(1):8-13.Nan Jun,Chai Yongming,Li Yanpeng,et al.Deactivation of Pd/Al2O3catalyst for selective hydrogenation of full-range reformate gasoline[J].Industrial Catalysis,2007,15(1):8-13.
[6]南军,柴永明,李彦鹏,等.UDO-01重整生成油选择性加氢催化剂的研制[J].石油炼制与化工,2007,38(1):28-33.Nan Jun,Chai Yongming,Li Yanpeng,et al.Preparation of UDO-01catalyst for selective hydrogenation of olefin in reformate[J].Petroleum Processing and Petrochemicals,2007,38(1):28-33.
[7]唐博合金,徐菁利.重整后加氢 Pd/Al2O3型催化剂的制备[J].石油与天然气化工,2010,39(1):32-34.Tang Bohejin,Xu Jingli.Preparation of reformed hydrogenation catalyst of Pd/Al2O3[J].Chemical Engineering of Oil &Gas,2010,39(1):30-34.
[8]Sales E A,Jove J,Mendes M,et al.Liquid-phase selective hydrogenation of hexa-1,5-diene and hexa-1,3-diene on Pd catalysts effect of Sn and Ag addition[J].Journal of Catalysis,2000,195(1):96-105.
[9]Bertolini J C,Jugnet Y.Surface structure and catalytic reactivity of palladium overlayers for 1,3-butadiene hydrogenation[J].The Chemical Physics of Solid Surfaces,2002,10:404-437.
[10]Gilles Berhault,Laure Bisson,Ce'cile Thomazeau,et al.Preparation of nanostructured Pd particles using a seeding synthesis approach-Application to the selective hydrogenation of buta-1,3-diene[J].Applied Catalysis A:General,2007,327(1):32-43.
[11]Vignéa F,Haubrich J,Loffreda D.Highly selective hydrogenation of butadiene on Pt/Sn alloy elucidated by first-principles calculations[J].Journal of Catalysis,2010,275(1):129-139.
[12]Jugnet Y,Sedrati R,Bertolini J C.Selective hydrogenation of 1,3-butadiene on Pt3Sn(Ⅲ)alloys:comparison to Pt(Ⅲ)[J].Journal of Catalysis,2005,229(1):252-258.
[13]李克明,冷家厂,王雨勃,等.分子筛催化剂脱除重整油中微量烯烃的研究[J].化学工业与工程,2009,26(5):429-432.Li Keming,Leng Jiachang,Wang Yubo,et al.Zeolite catalysts for removal of trace olefins in reformate without the presence of hydrogen[J].Chemical Industry and Engineering,2009,26(5):429-432.
[14]戴丹,王海彦,魏民,等.在 Ni-Mo/Al2O3上催化裂化轻汽油的选择性加氢[J].辽宁石油化工大学学报,2005,25(2):36-38.Dai dan,Wang haiyan,Wei min,et al.Selective hydrogenation of FCC light gasoline over Ni-Mo/Al2O3[J].Journal of Liaoning University of Petroleum & Chemical Technology,2005,25(2):36-38.
[15]Bachiller B B,Rodriguez R I,Guerrero-Ruiz A.Influence of Mg and Ce addition to ruthenium based catalysts used in the selective hydrogenation ofα,β-unsaturated aldehydes[J].Applied Catalysis A:General,2001,205(1/2):227-237.
[16]Inst Francais du Petrole.Method of selective hydrogenation of diolefin in steam cracking gasoline in the presence of catalyst based on carrier metal into which organic compound has been incorporated before being fed to reactor[P].Jpn.,JP4226592A,1992.
[17]中国石油化工股份有限公司.一种硫化型加氢催化剂的制备方法[P].中国:CN1861260A,2006.Sinopec Group.A sulfurized hydrogenation catalyst preparation method[P].China:CN1861260A,2006.
[18]梁顺琴,吴杰,王廷海,等.裂解汽油一段镍基加氢催化剂 LY-2008[J].石油科技论坛,2013,1:45-47.Liang Shunqin,Wu Jie,Wang Tinghai,et al.LY-2008Ni-Based catalyst for 1st pyrolysis gasoline hydrogenation[J].Oil Forum,2013,1:45-47.
[19]Wang Lu,Zhang Yongna,Zhang Yuliang,et al.Hydrodesulfurization of 4,6-DMDBT on a multi-metallic sulfide catalyst with layered structure[J].Applied Catalysis A:General,2011,394(1/2):18-24.
猜你喜欢
石油炼制与化工(2022年6期)2022-06-21 11:20:38
航空维修与工程(2022年11期)2022-02-06 06:37:28
天津医科大学学报(2021年1期)2021-12-05 11:11:05
工业催化(2020年9期)2020-11-13 08:20:36
储能科学与技术(2019年2期)2019-03-08 09:26:04
中国新技术新产品(2017年3期)2017-03-07 09:02:42
化工管理(2016年33期)2016-12-22 06:51:16
现代检验医学杂志(2016年5期)2016-08-20 03:17:08
山西化工(2016年6期)2016-04-09 07:17:41
茶叶通讯(2014年2期)2014-02-27 07:55:40
