
HPLC测定骨松宝颗粒中淫羊藿苷和朝藿定C的含量
2014-09-03 08:45:18穆建国甘惠仍贾宪生
中国合理用药探索 2014年6期
穆建国 甘惠仍 贾宪生
(1贵州富华药业有限责任公司,贵州 黔南州 551206;2贵阳中医学院,贵阳550002)
骨松宝颗粒是我公司独家生产的纯中药制剂(批准文号:国药准字Z52020006),药物组成为淫羊藿、续断、川芎、赤芍、知母、生地黄、莪术、三棱和牡蛎等,具有补肾活血、强筋壮骨的功效,临床用于骨瘘(骨质疏松)引起的骨折、骨痛、骨关节炎,以及预防更年期骨质疏松等症。骨松宝颗粒处方收载于《卫生部药品标准(中药成方制剂第十七册)》(WS-B-3292-98),原质量标准未制定含量测定项目,难以满足对产品质量控制的需要。本研究选择处方中君药淫羊藿所含较多的药理活性成分异戊烯基取代的黄酮类化合物淫羊藿苷和朝藿定C[1]作为指标成分,采用高效液相色谱法(HPLC)对骨松宝颗粒中淫羊藿苷和朝藿定C的含量进行测定[2],用以评价主药淫羊藿含量,该方法简便、快速、准确,重复性好,为有效提高骨松宝颗粒质量标准控制产品的质量提供了依据。
1 仪器与试药
1.1 仪器
岛津LC-10ATvp高效液相色谱仪(二元泵,SPD-10Avp紫外检测器,WML-2010色谱工作站);CX-250型超声波清洗器(功率 300W);AE-240电子分析天平[梅特勒-托利多仪器(上海)有限公司]。
1.2 试药
对照品:淫羊藿苷(原中国药品生物制品检定所提供,含量测定用,批号:110737-200415),朝藿定C(原中国药品生物制品检定所提供,含量测定用,批号:110780-200801);甲醇为色谱纯;水为高纯水,其他试剂为分析纯;骨松宝颗粒:贵州富华药业有限责任公司提供(含糖型,批号20110106)。
2 方法与结果
2.1 对照品溶液的制备
精密称取对照品朝藿定C和淫羊藿苷,加适量甲醇制成浓度分别为 1.076,0.020 8 mg/mL的溶液,作为对照品溶液。
2.2 供试品溶液的制备
取供试品,混匀,研细,取约2 g,精密称定,置具塞锥形瓶中,精密加入甲醇20 mL,密塞,称定重量,超声处理 1 h,放冷至室温,再称定重量,用甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。
2.3 色谱条件
色谱柱:大连依利特分析仪器有限公司Hypersil ODS2 (250 mm × 4.6 mm,5 μm);柱温:25 ℃;流动相:乙腈-0.1%磷酸溶液(25∶75)为流动相;流速1 mL/min;检测波长 270 nm;进样量:10 μL。
2.4 测定方法
分别精密吸取对照品溶液与供试品溶液各10 μL,注入高效液相色谱仪,测定对照品与供试品峰面积,计算。按上述色谱条件获得对照品和样品的色谱图见图1,2。
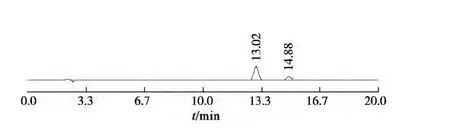
图1 朝藿定和淫羊藿苷混合对照色谱图(左为朝藿定C,右为淫羊藿苷)
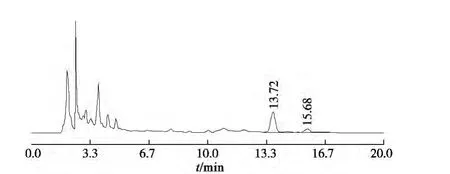
图2 供试品色谱图
2.5 线性关系考察
精密称取对照品朝藿定C淫羊藿苷,加适量甲醇制成其浓度分别约为 1.076,1.039 mg/mL的溶液,作为单一成分对照品储备溶液。精密吸取朝藿定的对照品溶液C 2.0 mL和淫羊藿苷的对照品溶液0.4 mL,置10 mL容量瓶中,加甲醇定容至刻度,摇匀,即得到每1 mL含朝藿定C和淫羊藿苷分别为0.215 2和0.041 6 mg的混合对照品溶液,精密吸取混合对照液 1,2,4,5,8,10,15,20 μL分别进样,按上述色谱条件测定,记录峰面积。以峰面积积分值(Y)为纵坐标、朝藿定C和淫羊藿苷对照品的量(X)为横坐标,绘制标准曲线。得朝藿定C近似过原点的线性回归方程:
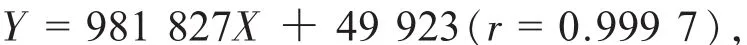
线性范围为 0.216~ 4.303 μg;得淫羊藿苷近似过原点的线性回归方程:
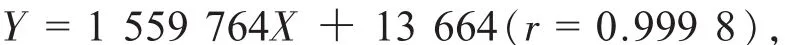
线性范围为0.042~ 0.831 μg。线性测定结果见表1。
2.6 精密度试验
取骨松宝颗粒剂适量混匀,研细,取2 g,精密称定,按含量测定项下方法制备供试液,连续进样9次,测得朝藿定C峰面积平均值1 431 761,RSD=0.13%,淫羊藿苷峰面积平均值260 823,RSD=0.99%,表明精密度良好。
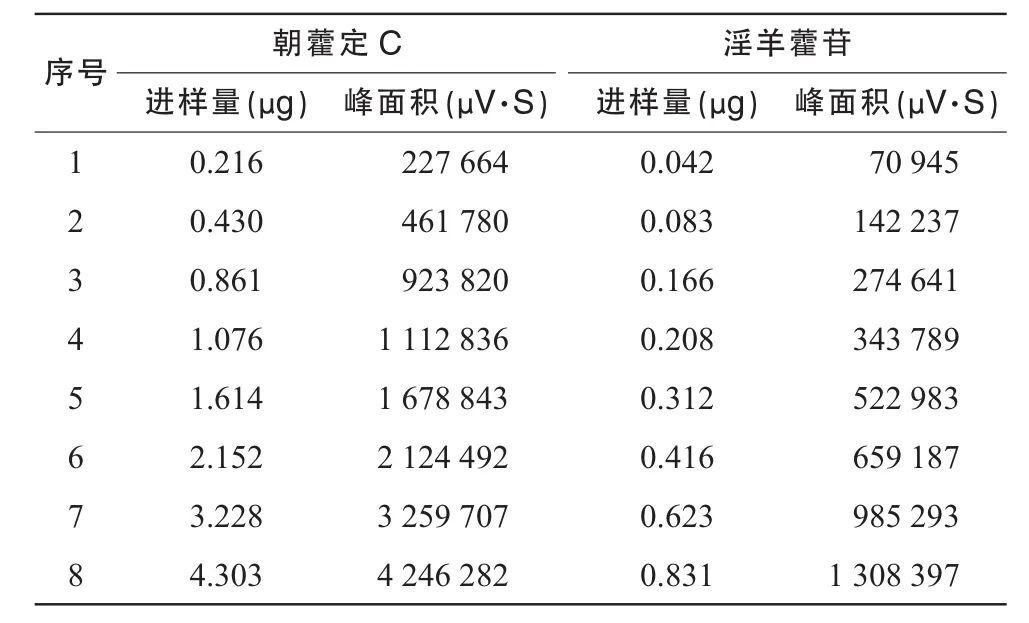
表1 朝藿定C和淫羊藿苷线性测定结果
2.7 重复性试验
取骨松宝颗粒适量混匀,研细,分别取6份,各2 g,精密称定,按质量标准草案含量测定项制备供试液,精密吸取供试液进样,计算朝藿定C含量和淫羊藿苷含量,朝藿定C含量平均值为1.477 mg/g,RSD=1.64%;淫羊藿苷含量平均值为0.166 mg/g,RSD=1.73%,表明重复性良好。
2.8 稳定性试验
取骨松宝颗粒适量混匀,研细,取2 g,精密称定,按含量测定项下方法制备供试液,精密吸取10 μL,在时间间隔为 0,2.0,4.0,6.0,8.0,10.0 h,分别进样供试品朝藿定C峰面积平均值1 337 801,RSD=1.98%,淫羊藿苷峰面积平均值 248 486,RSD=1.89%,结果表明本试验方法在10 h内稳定。
2.9 回收率试验
取骨松宝颗粒适量,混匀,研细,分别取9份,各1 g,精密称定,于1,2,3号样中分别加入朝藿定C对照品溶液(C=1.076 mg/mL)0.7 mL和淫羊藿苷对照品溶液(C=0.103 9 mg/mL)0.7 mL,相当于0.753 2 mg朝藿定C对照品和0.072 7 mg淫羊藿苷对照品;于4,5,6号样中分别加入朝藿定C对照品溶液(C=1.076 mg/mL)1.4 mL和淫羊藿苷对照品溶液(C=0.103 9 mg/mL)1.5 mL,相当于1.506 4 mg朝藿定C对照品和0.155 9 mg淫羊藿苷对照品;于7,8,9号样中分别加入朝藿定C对照品溶液(C=1.076 mg/mL)2.1 mL和淫羊藿苷对照品溶液(C=0.103 9 mg/mL)2.2 mL,相当于2.259 6 mg朝藿定C对照品和0.228 6 g淫羊藿苷对照品。按质量标准草案含量测定项制备供试液,进样,计算朝藿定C和淫羊藿苷回收率,朝藿定C回收率为98.53%~ 101.92%,RSD=0.98%,淫羊藿苷97.32%~103.69%,RSD=1.98%,见表2。

表2 加样回收试验测定结果(n=9)
2.10 样品含量测定
取10批骨松宝颗粒(含糖型),按2.2项下方法制备供试品溶液,按上述色谱条件测定,计算含量,结果见表3。
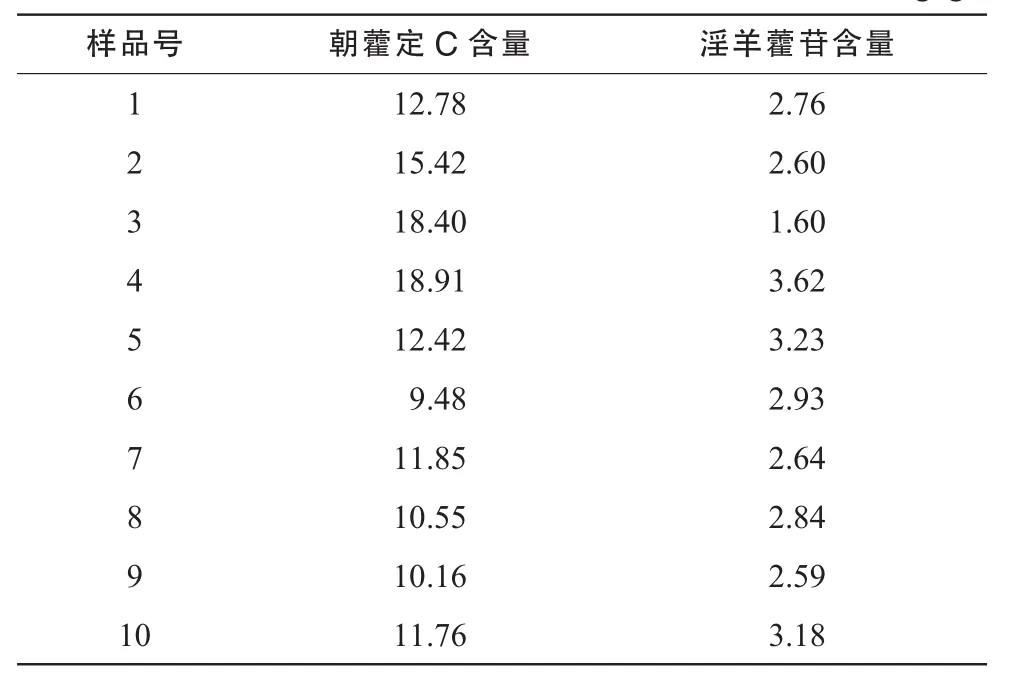
表3 10批骨松宝颗粒中朝霍定C和淫羊藿苷的含量测定结果 (mg/g)
3 讨论
3.1 检测波长的确定
骨松宝颗粒中含量测定的指标成分朝藿定C和淫羊藿苷为黄酮类成分,在265~ 275 nm和340~350 nm均有吸收。经对本品色谱分析检测波长 220,260,270,340 nm 的考察,在 270 nm 波长处朝藿定C和淫羊藿苷吸收峰灵敏度较高,其他色谱峰干扰相对较小,色谱峰的分离最为理想,故选用270 nm作为检测波长。
3.2 色谱柱的选择
分别考察了EliteHypersilODS2(5μm,250mm×4.6 mm)色谱柱和 Diamonsil(钻石)C18(5 μm,250 mm ×4.6mm)色谱柱,结果两个厂牌均可,Diamonsil色谱柱出峰时间较长,故选择前者。
3.3 流动相的选择
考察了甲醇-1%冰醋酸,乙腈-水,乙腈-0.1%磷酸等组成的流动相分离情况,以乙腈-0.1%磷酸系统较好;又考察了不同比例乙腈-0.1%磷酸(24∶76,25∶75,26∶74,28∶72)为流动相的分离情况,以乙腈-0.1%磷酸比例为 25∶75和 26∶74对两个成分的综合分离效果较好,后者出峰时间稍短,故确定以前者乙腈-0.1%磷酸(25∶75)为流动相比例;流速考察了 1.2,1.0,0.8 mL/min,除柱压和保留时间有较大差异外分离结果区别不大,1.0和 0.8 mL/min流速分离较1.2 mL/min流速分离稍好,故确定为1 mL/min。
3.4 指标性成分的确定
淫羊藿作为处方中君药,其主要活性成分为黄酮类化合物[3-4],其中朝藿定C含量较高[5],《中国药典》(2010年版)除巫山淫羊藿以朝藿定C外其余品种仅以淫羊藿苷作为药材质量控制标准[6],不能客观地反映和评价淫羊藿药材的内在质量[7],故考虑选择淫羊藿苷和朝霍定C作为质量控制指标。
本方法简便、快速、准确,重复性好,能较全面地控制骨松宝颗粒的质量,在实际使用中,为便于评价,可以将骨松宝颗粒中朝藿定C和淫羊藿苷的含量之和用以衡量制剂中淫羊藿含量进而作为骨松宝颗粒含量的质量控制指标。本研究结果为骨松宝颗粒质量标准的提升提供了依据。
[1]孟宁,孔凯,李师翁.淫羊藿属植物化学成分及药理活性研究进展[J].西北植物学报,2010,30(5):1063-1073.
[2]李晓龙,刘虹宇,曹佩雪,等.HPLC同时测定21种淫羊藿中朝藿定 C和淫羊藿苷的含量[J].药物分析杂志,2011,31(5):137-140.
[3]张玉萱,徐玲玲.淫羊藿总黄酮的药理作用研究进展[J].实用临床医药杂志,2012,16(9):125-128.
[4]蔡曼玲,季晖,李萍,等.5种淫羊藿黄酮类成分对体外培养成骨细胞的影响[J].中国天然药物,2004,2(4):235-238.
[5]雷永涛,梁妍,郝小燕,等.不同产地淫羊藿中四种活性成分含量的高效液相色谱法测定[J].时珍国医国药,2013,24(6):1404-1405.
[6]国家药典委员会.中华人民共和国药典(2010年版)一部[S].北京:中国医药科技出版社,2010:155,306.
[7]郭宝林,肖培根.中药淫羊藿主要种类评述[J].中国中药杂志,2003,28(4):303-307.
猜你喜欢
食品与发酵工业(2023年21期)2023-11-26 07:50:24
江西中医药(2022年8期)2022-08-22 02:01:26
煤化工(2022年3期)2022-07-08 07:24:42
中成药(2018年12期)2018-12-29 12:26:00
中国中医药现代远程教育(2018年22期)2018-02-09 02:12:04
湖南林业科技(2017年6期)2018-01-30 03:48:06
中成药(2017年4期)2017-05-17 06:09:49
中国资源综合利用(2016年10期)2016-01-22 08:36:09
陕西中医(2015年11期)2015-03-22 04:29:17
郑州大学学报(理学版)(2014年2期)2014-03-01 04:20:54
