
水溶液中[Zn (NH3)m(H2O)n]2+(m+n=4)配合物结构和性质的DFT研究
2014-09-01 02:36尹周澜丁治英胡久刚陈启元
湖南师范大学自然科学学报 2014年4期
陈 静,尹周澜,丁治英,胡久刚,陈启元
(中南大学化学化工学院,中国 长沙 410083)
水溶液中[Zn (NH3)m(H2O)n]2+(m+n=4)配合物结构和性质的DFT研究
陈 静,尹周澜,丁治英,胡久刚,陈启元*
(中南大学化学化工学院,中国 长沙 410083)
采用密度泛函B3LYP方法,对水溶液中四面体配合物[Zn (NH3)m(H2O)n]2+(m+n=4) 结构和性质进行研究.在优化几何构型基础上,计算其最稳定构象的结合能及振动频率.结果表明:随着H2O逐渐被NH3取代,配合物中的Zn2+电荷主要转移到取代H2O的NH3中的H上;Zn-O键和Zn-N键的键长逐渐增加,Zn-O键长始终大于Zn-N键长;随着m增大,配合物结合能变大; NH3数目的增加将导致Zn-O和Zn-N振动向低频方向移动,而O-H键和N-H键的各种振动谱线则会缓慢蓝移.
密度泛函;电荷分布;结合能;振动频率
“氨浸-萃取-酸性电积”工艺[1-3]因具有可处理原料范围广,净化负担小,工艺流程短,环境污染小等优点而越来越多得受到人们的青睐.但该工艺用于锌湿法冶金体系时,由于其锌萃取率不高,限制了整个工艺在锌冶金中的发展.因此,氨浸体系中关于锌萃取机理的研究显得至关重要,而了解氨浸溶液中的物种结构及其性质是研究其萃取机理的基础.
目前对氨性溶液中Zn(II)-NH3-H2O配合物种类、结构和性质的研究还十分有限.胡久刚等[4]采用XANES光谱和EXAFS光谱等检测技术,发现低pH值下Zn(II)-NH3-H2O体系中配位结构为八面体的六配位结构和四面体的四配位构型,随着pH值增加,八面体构型的锌物种的相对含量急剧减少;EXAFS光谱拟合结果显示,Zn-O键长和Zn-N键长分别为2.08±0.02 Å和2.03±0.02 Å.在采用量子化学理论方法研究方面,Fatmi等[5-10]通过QM/MM MD方法对几种不同的Zn(II)-NH3-H2O配合物的结构及其动力学性质进行了探究,但已研究的四配位结构仅有 [Zn(NH3)4]2+.因此,研究不同四配位[Zn (NH3)m(H2O)n]2+(m+n=4)配合物的结构特征,可了解氨浸溶液中的物种结构及其性质,从而为进一步探究氨性溶液中二价锌离子的萃取机理提供理论基础.
本文采用量子化学计算方法对液相体系中所有的[Zn (NH3)m(H2O)n]2+(m+n=4)四配位配合物结构特征进行研究,通过优化其基态结构,计算配合物离子的结合能,并对NBO布居和光谱性质进行分析.
1 计算方法
Dunning的双-zeta DZP基组可以成功应用于H、N和O原子的量子化学计算 ,增加弥散函数的DZP diffuse基组可以得到更加准确的值.而在过渡金属配合物的结构计算中,适用于过渡金属的Stuttgart/Dresden ECP基组(标识为SDD)可得到相对较好的结构数据.
在整个计算过程中,采用Becke梯度修正交换函数和Lee-Yang-Parr与Vosko-Wilk-Nusai带有部分额外HF交换能相关函数的混合参数泛函B3LYP方法(对H、N和O使用DZP diffuse基组,对Zn使用SDD基组),采用IEF-PCM溶剂模型在液相条件下完成所有的结构优化、频率计算和NBO布居分析.
在已优化结构的基础上采用MP2/6-311++G(2d,2p)方法计算[Zn (NH3)m(H2O)n]2+(m+n=4) 稳定结构的结合能,结合能的计算方法如式(1)所示:
-△E=E(Zn[(NH3)m(H2O)n]2+)(corrected)-mE(NH3)-nE(H2O)
其中,E(Zn[(NH3)m(H2O)n]2+)(corrected)、E(NH3)和E(H2O)分别表示经过BSSE校正后的配合物能量、单个NH3和单个H2O的能量.由于Zn2+、NH3和H2O的基函数在配合物中重叠,从而增大了配合物的基组而使-△E能量降低,这部分贡献如果也掺入-△E,则将会高估结合能,所以需要通过BSSE校正去掉高估的部分.
整个计算采用Gaussian03[17]计算程序,计算过程在中南大学高性能计算平台上完成.
2 结果与讨论
2.1 NBO布居分析
表1列出了自由H2O分子、NH3分子和[Zn (NH3)m(H2O)n]2+(m+n=4)四配位配合物中所有原子的NBO电荷分布.

表1 自由H2O分子、NH3分子和[Zn (NH3)m(H2O)n]2+(m+n=4)配合物电荷分布
从表1中数据可以看出,与自由H2O分子、NH3分子相比,由于Zn2+离子表面电荷的转移,[Zn (NH3)m(H2O)n]2+(m+n=4)四配位配合物中H2O分子、NH3分子所有原子表面的电荷均出现不同程度的增加,这表明水化作用的结果使Zn2+电荷向H2O分子和NH3分子上的原子转移.而随着NH3分子数目的增加,四配位配合物中所有原子上的电荷的变化均呈现出明显的规律性,O原子和N原子上的电荷略微增加,而O原子和N原子上H原子的电荷却略微减小.与H、N和O原子上电荷的微小变化相比,Zn离子表面的电荷减小较为明显,这是由于随着2个H的H2O分子被3个H的NH3分子取代,增加了H原子的个数,而Zn2+上的电荷也正逐渐转移到了NH3分子中增加的H上,这表明N的作用使Zn2+电荷逐渐向NH3上的H转移.
2.2 几何构型
[Zn (NH3)m(H2O)n]2+(m+n=4)不同配合物的优化结构如图1所示.
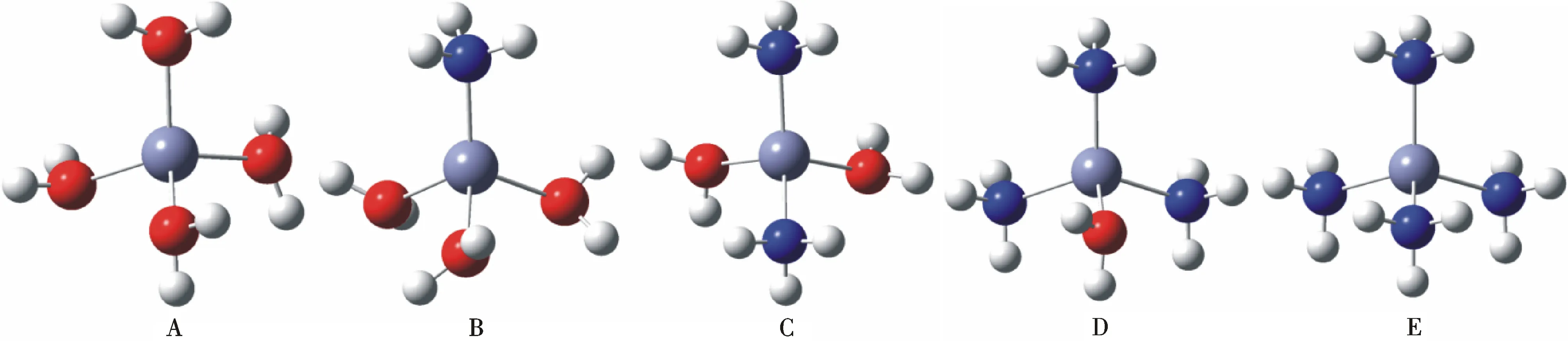
图1 [Zn (NH3)m(H2O)n]2+(m+n=4)不同配合物的优化结构Fig.1 Optimized geometries of [Zn (NH3)m(H2O)n]2+(m+n=4) complexes

图2 [Zn (NH3)m(H2O)n]2+(m+n=4)不同配合物键长随氨配位数增加的变化(单位:Å)Fig.2 Bond lengths of [Zn (NH3)m(H2O)n]2+(m+n=4) complexes (Unit: Å)
图2表示[Zn (NH3)m(H2O)n]2+(m+n=4)不同配合物Zn-O键长、Zn-N键长随氨配位数增加的变化.
从图2中可以明显的看出,随着NH3分子逐渐取代[Zn(H2O)4]2+配合物中的H2O分子,Zn-O键长和Zn-N键长逐渐增大,2.1中NBO布居分析表明这是由于Zn和N、O原子表面相对电荷减小,静电力减弱所致;而且,Zn与N之间的相对电荷明显大于Zn与O,导致N与Zn有着比O与Zn更大的亲和力,因此,在所有的[Zn (NH3)m(H2O)n]2+(m+n=4)的四配位配合物中,Zn-O键长大于Zn-N键长,Fatmi等[5-10]通过QM/MM MD模拟水溶液中[Zn(H2O)6]2+配合物的一、二、三氨取代物也发现了类似的结论.同时,Zn-O键长变化范围(1.98 Å~2.07 Å)和Zn-N键长变化范围(1.98 Å~2.05 Å)与胡久刚[4]通过EXAFS光谱拟合的结果较为一致.
表2列出了[Zn (NH3)m(H2O)n]2+(m+n=4)配合物O-Zn-O键角、O-Zn-N键角、N-Zn-N键角数据.

表2 [Zn (NH3)m(H2O)n]2+(m+n=4)配合物键角随氨配位数增加的变化(单位:゜)
表2中数据表明,随着NH3分子逐渐取代[Zn(H2O)4]2+配合物中的H2O分子,O-Zn-O键角、O-Zn-N键角和N-Zn-N键角几乎等比例减小.当[Zn (NH3)m(H2O)n]2+(m+n=4)四配位配合物中同时存在NH3分子和H2O分子时,由于N与Zn2+和O与Zn2+不同的亲和力导致整个配合物结构的不完全对称,O-Zn-O键角、O-Zn-N键角和N-Zn-N键角存在14°~18°左右的差距.但是,当NH3分子完全取代H2O分子后,N-Zn-N键角由于作用力的重新均衡又再次恢复到与[Zn(H2O)4]2+配合物中O-Zn-O键角完全相同的109.5°.与Fatmi等[7]通过QM/MM MD模拟的[Zn(H2O)4]2+配合物中N-Zn-N键角(109°)数据基本一致.
2.3 结合能
图3给出了[Zn (NH3)m(H2O)n]2+(m+n=4)不同配合物的结合能随NH3配位数的变化.

图3 [Zn (NH3)m(H2O)n]2+(m+n=4)不同配合物结合能随氨配位数增加的变化Fig.3 Binding energy of [Zn (NH3)m(H2O)n]2+(m+n=4) complexes
从图3中可以看出,由于Zn-N键能大于Zn-O键,随着NH3分子逐渐取代[Zn(H2O)4]2+配合物中的H2O分子,[Zn (NH3)m(H2O)n]2+(m+n=4)四配位配合物的总结合能逐渐增大.不过,随着NH3数目逐渐接近4,四配位配合物的的内部作用力逐渐恢复均衡,结合能增加的趋势逐渐减小.
2.4 振动频率
表3为随着[Zn (NH3)m(H2O)n]2+(m+n=4)配合物中的H2O分子逐渐被NH3分子取代时主要的振动频率.包括Zn-O键和Zn-N键伸缩振动,O-H键和N-H键的弯曲振动以及对称与不对称伸缩振动,其它振动由于不明显或者两种或两种以上振动混合在一起而未被列出.同时,考虑DFT模型和SDD/DZP基组引入了一定的计算误差,所有结果均为乘以0.961 4频率矫正因子[12]后的数据.

表3 [Zn (NH3)m(H2O)n]2+(m+n=4)不同配合物中主要的振动频率(单位:cm-1)
表中数据表明,随着[Zn(H2O)4]2+配合物中的H2O分子逐渐被NH3分子取代,各种振动频率变化都存在一定的规律:由于Zn2+的极化作用减弱,使Zn-O和Zn-N振动向低频方向移动;而O-H键和N-H键的各种振动频率却出现完全相反的变化,均出现蓝移的现象.
3 结论
随着[Zn (NH3)m(H2O)n]2+(m+n=4)四配位配合物中的H2O分子逐渐被NH3分子取代,配合物中的Zn2+电荷主要转移到取代H2O的NH3分子中的H上;Zn-O键和Zn-N键的键长逐渐增大,Zn-O键稍长于Zn-N键,主要的键角如O-Zn-O键角、O-Zn-N键角和N-Zn-N键角几乎等比例的减小;随着NH3数目的增加,由于Zn-N键键能大于Zn-O键,从而导致配合物的结合能变大;此外,NH3数目的增加将导致Zn-O键和Zn-N键伸缩振动吸收光谱红移,而O-H键和N-H键的各种振动则缓慢蓝移.计算结果与我们的实验结果以及已知文献中的部分相关数据一致性良好.
[1] CHEN Q, LI L, BAI L,etal. Synergistic extraction of zinc from ammoniacal ammonia sulfate solution by a mixture of a sterically hinderedβ-diketone and tri-n-octylphosphine oxide(TOPO)[J]. Hydrometallurgy, 2010,105(3-4): 201-206.
[2] DING Z, YIN Z, HU H,etal. Dissolution kinetics of zinc silicate(hemimorphite) in ammoniacal solution[J]. Hydrometallurgy, 2010,104(2):201-206.
[3] YIN Z, DING Z, HU H,etal. Dissolution of zinc silicate(hemimorphite) with ammonia-ammonium chloride solution[J]. Hydrometallurgy, 2010,103(1-4):215-220.
[4] 胡久刚. 氨性溶液中铜、镍、锌金属离子的萃取行为及微观机理的研究[D]. 长沙: 中南大学, 2012.
[5] FATMI M Q, HOFER T S, RODE B M. The stability of [Zn(NH3)4]2+in water: A quantum mechanical/molecular mechanical moleculer dynamics study[J]. Phys Chem Phys, 2010,12(33):9713-9718.
[6] FATMI M Q, HOFER T S, RANDOLF B R,etal. Temperature effects on the structural and dynamics properties of the Zn(II)-water complex in aqueous solution: A QM/MM molecular dynamics study [J]. J Phy Chem B, 2005,110(1):616-621.
[7] FATMI M Q, HOFER T S, RANDOLF B R,etal. An extended ab initio QM/MM MD approach to structure and dynamics of Zn(II) in aqueous solution [J]. J Chem Phys, 2005,123(054514):4514.
[8] FATMI M Q, HOFER T S, RODE B M,etal. Structure and dynamics of [Zn(NH3)(H2O)5]2+complex in aqueous solution obtained by an ab initio QM/MM molecular dynamics study [J]. Phys Chem Phys, 2006, 8(14):1675-1681.
[9] FATMI M Q, HOFER T S, RANDOLF B R,etal. Stability of different Zinc(II)-diamine complexes in aqueous solution with respect to structure and dynamics: A QM/MM MD study [J]. J Phys Chem B, 2007,111(1):151-158.
[10] FATMI M Q, HOFER T S, RANDOLF B R,etal. Exploring structure and dynamics of the diaquotriamminezinc(II) complex by QM/MM MD situlation [J]. J Phys Chem B, 2008,112(18):5788-5794.
[11] FRISCH M J, TRUCKS G W, SCHLEGEL H B,etal. Gaussian 03, Revision E. 01, Gaussian, Inc., Pittsburgh PA, 2004.
[12] GALABOV B, YAMAGUCHI Y, REMINGTON R B,etal. High level ab initio quantum mechanical predictions of infrared intensities[J]. J Phys Chem A, 2002,106(5):819-832.
(编辑 杨春明)
A DFT Study of Structure and Properties for
[Zn(NH3)m(H2O)n]2+(m+n=4) in Aqueous Solution
CHENJing,YINZhou-lan,DINGZhi-ying,HUJiu-gang,CHENQi-yuan*
(College of Chemistry and Chemical Engineering, Central South University, Changsha 410083, China)
The 4-coordinate complexes [Zn (NH3)m(H2O)n]2+(m+n=4) in aqueous solution were investigated by density function theory calculations (B3LYP functional). The geometries for the complexes were optimized to obtain the lowest-energy structures, of which the binding energies and vibrational frequencies were then calculated. The results show that the charge of zinc ions is mainly transferred to the hydrogen atoms of ammonia molecules which have taken the place of water molecules. The Zn-O bond length and Zn-N bond length increase gradually when water molecules are replaced by ammonia molecules, and the Zn-O bond is always longer than the Zn-N bond. With the increase ofm, the binding energy of complexes becomes larger. As the number of ammonia molecules increases, the vibration of Zn-O and Zn-N bonds trends to lower frequencies. Meanwhile, the vibrational spectra of O-H bond and N-H bond will have a little blue-shift.
density function theory; charge distribution; binding energy; vibrational frequency
2014-03-26
国家重点基础研究发展计划资助项目(973计划)(2014CB643401);自然科学基金重点资助项目(51134007)
*
,E-mail:xhli@csu.edu.cn
TF111
A
1000-2537(2014)04-0038-04
猜你喜欢
大学物理(2022年9期)2022-09-28
高中数理化(2022年16期)2022-09-14
吉林大学学报(理学版)(2021年1期)2021-01-18
物理通报(2020年7期)2020-07-01
青岛大学学报(工程技术版)(2019年2期)2019-09-10
中学生数理化(高中版.高考理化)(2019年6期)2019-06-22
火工品(2019年6期)2019-06-05
厦门大学学报(自然科学版)(2018年2期)2018-04-11
当代化工研究(2016年5期)2016-03-20
原子与分子物理学报(2015年3期)2015-11-24
