
空气氧化法制备3-羟基季碳氧化吲哚
2014-08-13 01:39:00郭丰敏刘雄伟刘雄利余章彪
遵义医科大学学报 2014年4期
郭丰敏,杨 超,刘雄伟,周 英,刘雄利,余章彪
(贵州大学 药学院暨贵州省中药民族药创制工程中心,贵州 贵阳 550025)
氧气由于其广泛存在于空气中,成本极低,并且由于其几乎没有污染的副产物产生,因此被认为是最理想的氧化剂。在最近几十年里,过渡金属催化的有机底物的空气氧化法已经有文献报道[1]。特别是官能团α-位的氧原子插入可以构建碳氧键[2],在药物分子的官能团合成中具有重要的意义。
3-羟基季碳氧化吲哚普遍存在于生物活性的天然产物和药物分子中[3-5], 也是作为合成天然生物碱的起始原料或中间体[6-8],例如,Convolutamydines A-E[9-12]是一类从海洋生物中提取出来的生物碱,它们的主体结构是含有一个季碳的3-羟基氧化吲哚,个体不同体现在C3位上的不同取代,目前研究已经发现它们对不同的细胞具有很好的响应性。通过文献调研发现,合成3-羟基季碳氧化吲哚目前主要是采用金属作为催化剂或者直接用金属试剂做原料对靛红衍生物进行亲核加成反应。本文通过空气做氧化剂,没有金属催化剂或金属试剂参与,直接采用3-取代氧化吲哚在碳酸钾和相转移催化剂TBAB条件下,高效合成8个未见文献报道的3-季碳羟基氧化吲哚(2a~2h),结构经1H NMR,13C NMR和HR MS表征。
1 材料与方法
1.1 仪器与试剂 Bruker-300 MHz 型核磁共振仪(CDCl3为溶剂,TMS为内标); Bruker BIO TOF III Q 型高分辨质谱仪。所用试剂均为分析纯;所用无水溶剂均按标准程序进行脱水处理。
1.2 目标化合物2a的合成 在反应管中依次加入267 mgN-甲基-3-对甲氧基苄基取代氧化吲哚(1.0 mmol)、32.2 mg 四丁基溴化铵(TBAB, 0.1mmol,10mol%) 和13.8mgK2CO3(0.1mmol, 10mol%),5 mL甲苯,在空气气氛中室温下搅拌反应48 h,TLC检测至反应完成后,直接上硅胶层析柱,乙酸乙酯/石油醚(V/V)1/4为洗脱剂,柱层析纯化得到254.7 mg 3-羟基-3-(4-甲氧基苄基)-1-甲基氧化吲哚(2a)。通过上述方法,相应反应底物按同样计量比投量,在空气气氛中室温下搅拌反应48 h,分别合成2b~2h,其物理常数和高分辨质谱分析见表1。
表1化合物2a~2h的物理常数及高分辨分析数据
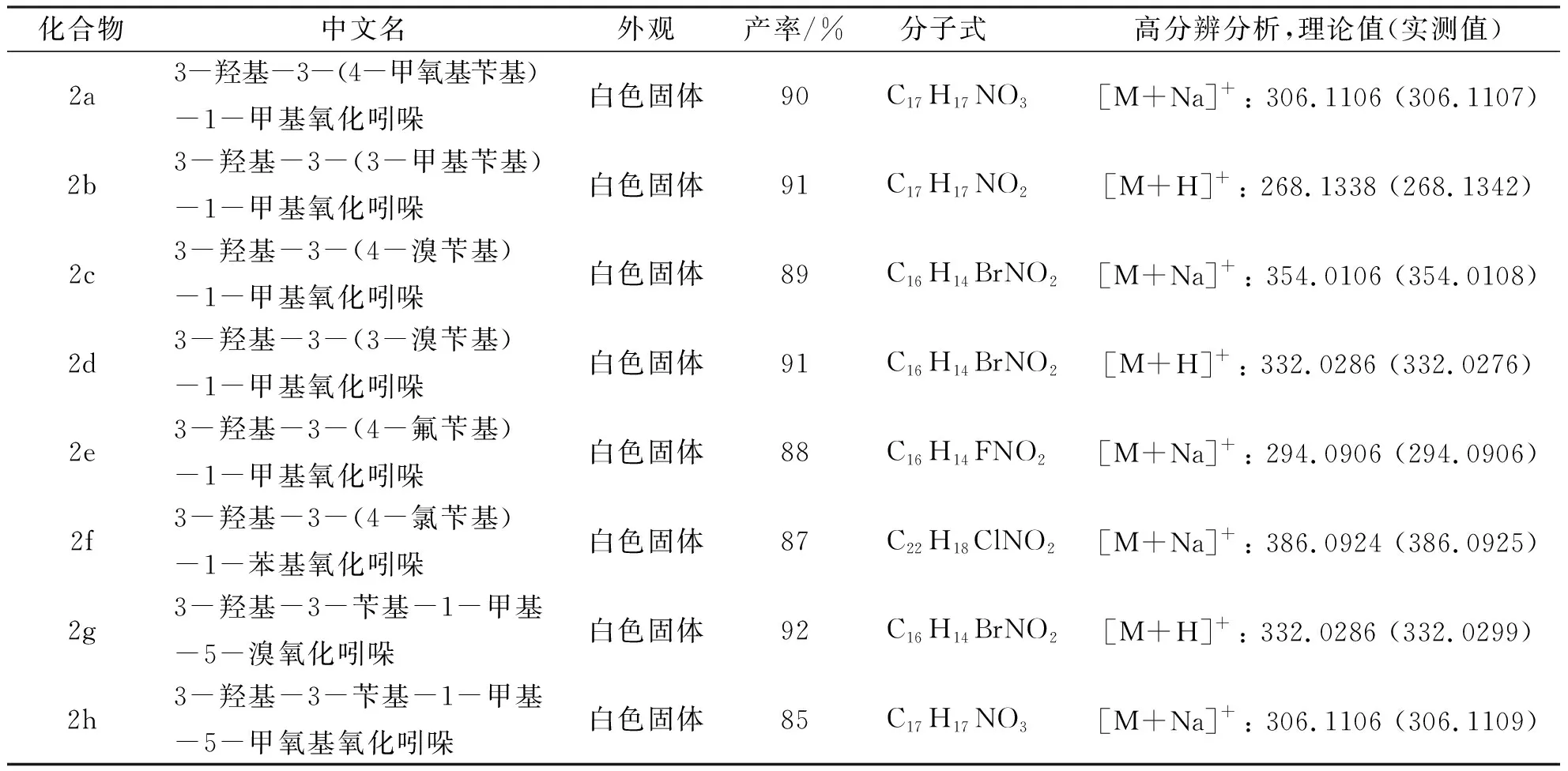
化合物中文名外观产率/%分子式 高分辨分析,理论值(实测值)2a3-羟基-3-(4-甲氧基苄基)-1-甲基氧化吲哚白色固体90C17H17NO3 [M+Na]+: 306.1106 (306.1107)2b3-羟基-3-(3-甲基苄基)-1-甲基氧化吲哚白色固体91C17H17NO2[M+H]+: 268.1338 (268.1342)2c3-羟基-3-(4-溴苄基)-1-甲基氧化吲哚白色固体89C16H14BrNO2 [M+Na]+: 354.0106 (354.0108)2d3-羟基-3-(3-溴苄基)-1-甲基氧化吲哚白色固体91C16H14BrNO2[M+H]+: 332.0286 (332.0276)2e3-羟基-3-(4-氟苄基)-1-甲基氧化吲哚白色固体88C16H14FNO2 [M+Na]+: 294.0906 (294.0906)2f3-羟基-3-(4-氯苄基)-1-苯基氧化吲哚白色固体87C22H18ClNO2 [M+Na]+: 386.0924 (386.0925)2g3-羟基-3-苄基-1-甲基-5-溴氧化吲哚白色固体92C16H14BrNO2 [M+H]+: 332.0286 (332.0299)2h3-羟基-3-苄基-1-甲基-5-甲氧基氧化吲哚白色固体85C17H17NO3 [M+Na]+: 306.1106 (306.1109)
2 目标化合物的结构表征
2a:1H NMR (CDCl3, 300 MHz)δ:2.91 (s, 3H, 1-CH3), 3.34 (d,J= 13.5 Hz, 1H, 1′-benzyl-CH2), 3.62 (s, 3H, 4′-benzyl-OCH3), 3.47 (d,J= 13.5 Hz, 1H, 1′-benzyl-CH2), 6.09 (br s, 1H, 3-OH), 6.53 (t,J= 7.2 Hz, 3H), 6.75 (d,J= 8.1 Hz, 2H), 6.99 (t,J= 7.2 Hz, 1H), 7.13 (t,J= 7.2 Hz, 1H), 7.22 (d,J= 7.5 Hz, 1H);13C NMR (CDCl3, 75 MHz)δ: 25.8, 35.2, 55.1, 76.5, 107.3, 112.8, 121.5, 124.2, 127.5, 128.1, 130.5, 131.1, 144.2, 157.4, 178.7。
2b:1H NMR (CDCl3, 300 MHz)δ: 2.05 (s, 3H, 3′-benzyl-CH3), 2.95 (s, 3H, 1-CH3), 3.33 (d,J= 13.5 Hz, 1H, 1′-benzyl-CH2), 3.47 (d,J= 13.5 Hz, 1H, 1′-benzyl-CH2), 6.12 (br s, 1H, 3-OH), 6.55 (d,J= 7.2 Hz, 1H, 1′-benzyl-CH2), 6.61-6.64 (m, 2H), 6.80-6.88 (m, 2H), 7.01 (t,J= 7.2 Hz, 1H), 7.11-7.14 (m, 1H), 7.24 (d,J= 7.5 Hz, 1H);13C NMR (CDCl3, 75 MHz)δ: 21.1, 25.8, 36.3, 76.6, 107.2, 121.3, 124.1, 126.8, 126.9, 127.0, 127.4, 130.7, 131.1, 135.8, 136.4, 144.1, 178.7。
2c:1H NMR (CDCl3, 300 MHz)δ: 2.98 (s, 3H, 1-CH3), 3.32 (d,J= 13.2 Hz, 1H, 1′-benzyl-CH2), 3.39 (d,J= 13.2 Hz, 1H, 1′-benzyl-CH2), 5.97 (br s, 1H, 3-OH), 6.58 (d,J= 7.8 Hz, 1H), 6.70 (d,J= 8.1 Hz, 2H), 7.00 (t,J= 7.2 Hz, 1H), 7.09-7.18 (m, 3H), 7.24 (d,J= 7.2 Hz, 1H);13C NMR (CDCl3, 75 MHz)δ: 26.0, 35.6, 76.3, 107.7, 120.2, 121.5, 124.0, 127.7, 130.5, 130.6, 131.5, 135.0, 144.3, 178.5。
2d:1H NMR (CDCl3, 300 MHz)δ: 2.98 (s, 3H, 1-CH3), 3.34 (d,J= 13.5 Hz, 1H, 1′-benzyl-CH2), 3.40 (d,J= 13.5 Hz, 1H, 1′-benzyl-CH2), 6.01 (br s, 1H, 3-OH), 6.72 (d,J= 7.2 Hz, 1H), 6.79 (d,J= 7.2 Hz, 1H), 6.90-7.02 (m, 3H), 7.11-7.17 (m, 2H), 7.41 (d,J= 7.5 Hz, 1H);13C NMR (CDCl3, 75 MHz)δ: 25.8, 35.7, 76.4, 107.5, 120.4, 121.3, 124.2, 127.7, 128.4, 128.8, 129.4, 130.1, 132.2, 138.9, 144.2, 178.0。
2e:1H NMR (CDCl3, 300 MHz)δ: 2.91 (s, 3H, 1-CH3), 3.32 (d,J= 13.2 Hz, 1H, 1′-benzyl-CH2), 3.41 (d,J= 13.2 Hz, 1H, 1′-benzyl-CH2), 6.02 (br s, 1H, 3-OH), 6.72-6.89 (m, 4H), 7.11-7.17 (m, 2H), 7.21-7.24 (m, 1H), 7.60 (d,J= 7.5 Hz, 1H);13C NMR (CDCl3, 75 MHz)δ: 25.9, 35.5, 76.0, 114.5, 114.8, 115.0, 123.4, 124.2, 127.7, 128.7, 130.4, 130.4, 131.3, 131.4, 140.0, 160.2, 163.4, 177.8。
2f:1H NMR (CDCl3, 300 MHz)δ: 3.30 (d,J= 13.2 Hz, 1H, 1′-benzyl-CH2), 3.43 (d,J= 13.5 Hz, 1H, 1′-benzyl-CH2), 6.12 (br s, 1H, 3-OH), 6.57 (d,J= 16.2 Hz, 1H), 4.94 (d,J= 16.2 Hz, 1H), 6.35-6.39 (m, 1H), 6.70-6.76 (m, 2H), 6.90-6.94 (m, 2H), 7.00-7.18 (m, 7H), 7.31-7.37 (m, 1H);13C NMR (CDCl3, 75 MHz)δ: 35.5, 43.2, 76.7, 108.5, 121.1, 124.0, 126.3, 126.7, 126.8, 127.6, 127.7, 128.2, 130.0, 130.9, 135.5, 136.1, 143.8, 178.7。
2g:1H NMR (CDCl3, 300 MHz)δ: 2.92 (s, 3H, 1-CH3), 3.32 (d,J= 13.5 Hz, 1H, 1′-benzyl-CH2), 3.44 (d,J= 13.5 Hz, 1H, 1′-benzyl-CH2), 6.11 (br s, 1H, 3-OH), 6.42 (d,J= 8.1 Hz, 1H), 6.81-6.86 (m, 2H), 7.04-7.08 (m, 3H), 7.21-7.26 (m, 1H), 7.47 (d,J= 1.8 Hz, 1H);13C NMR (CDCl3, 75 MHz)δ: 25.8, 35.7, 76.7, 108.1, 114.0, 126.7, 127.4, 127.8, 129.7, 130.0, 133.1, 135.5, 143.6, 178.4。
2h:1H NMR (CDCl3, 300 MHz)δ: 2.92 (s, 3H, 1-CH3), 3.30 (d,J= 13.5 Hz, 1H, 1′-benzyl-CH2), 3.44 (d,J= 13.5 Hz, 1H, 1′-benzyl-CH2), 3.75 (s, 3H, 5-OCH3), 6.11 (br s, 1H, 3-OH), 6.43 (d,J= 7.8 Hz, 1H), 6.83-6.86 (m, 2H), 6.93 (d,J= 7.8 Hz, 1H), 7.00-7.05 (m, 4H);13C NMR (CDCl3, 75 MHz)δ: 25.8, 35.7, 55.8, 76.7, 107.0, 125.1, 126.4, 127.4, 127.7, 129.9, 130.7, 131.1, 136.4, 142.1, 178.7。
3 结果
以N-甲基-3-取代氧化吲哚为起始原料,通过空气做氧化剂,没有金属催化剂或金属试剂参与,直接在碳酸钾和相转移催化剂TBAB条件下,高效合成8个未见文献报道的3-季碳羟基氧化吲哚(2a~2h),合成产率在85%~92%。并采用1H NMR,13C NMR和HRMS等技术确定了产物结构。目标化合物合成路线(见图1)。

图1 3-羟基季碳氧化吲哚的合成路线
4 讨论
通过对底物的扩展,我们可以发现该反应无论氧化吲哚3-取代苄基苯环上是吸电子或给电子取代(2a~2f),还是间位或对位取代(2a~2d),都能在空气做氧化剂,碳酸钾和相转移催化剂TBAB条件下,高效合成3-羟基季碳氧化吲哚。此外,3-取代氧化吲哚吲哚环上为给电子或吸电子取代(2f~2g),对产率没有明显影响。该方法提供了一种高效合成3-羟基季碳氧化吲哚类化合物的方法,不仅反应条件温和,反应时间短,产率较高,而且实验操作简单,分离容易。
[参考文献]
[1] Punniyamurthy T, Subbarayan V, Javed I, et al. Recent Advances in Transition Metal Catalyzed Oxidation of Organic Substrates with Molecular Oxygen [J].Chem Rev,2005,105 (6):2329-2363.
[2] Christoffers J, Baro A, Werner T, et al. α-Hydroxylation of β-Dicarbonyl Compounds [J].Adv Synth Catal,2004,346(2):143-151.
[3] Tokunaga T, Hume W E, Nagamine J, et al. Structure-activity relationships of the oxindole growth hormone secretagogues [J]. Bioorg Med Chem Lett,2005,15(7):1789-1792.
[4] Tokunaga T, Hume W E, Umezome T, et al. Oxindole Derivatives as Orally Active Potent Growth Hormone Secretagogues [J]. J Med Chem,2001, 44(26): 4641-4649.
[5] Hawawasam P, Erway M, Moon S L, et al. Synthesis and Structure-Activity Relationships of 3-Aryloxindoles:A New Class of Calcium-Dependent, Large Conductance Potassium (Maxi-K) Channel Openers with Neuroprotective Properties [J]. J Med Chem,2002,45(7):1487-1499.
[6] Kitajima M, Mori I, Arai K, et al. Two new tryptamine-derived alkaloids from Chimonanthus praecox f. concolor [J]. Tetrahedron Lett, 2006, 47(19): 3199-3202.
[7] Sua′rez-Castillo O R, Sa′nchez-Zavala M, Mele′ndez-Rodríguez M, et al. Preparation of 3-hydroxyoxindoles with dimethyldioxirane and their use for the synthesis of natural products[J].Tetrahedron, 2006, 62(13): 3040-3051.
[8] Kawasaki T, Nagaoka M, Satoh T, et al. Synthesis of 3-Hydroxyindolin-2-one[J].Tetrahedron ,2004, 62(13): 3040-3051.
[9] Zhang H P, Shigemori H, Ishibashi M, et al. Convolutamides A~F, novel γ-lactam alkaloids from the marine bryozoan Amathia convolute[J].Tetrahedron, 1994, 50(34): 10201-10206.
[10] Kamano Y, Zhang H P, Ichihara Y, et al. Convolutamydine A, a novel bioactive hydroxyoxindole alkaloid from marine bryozoan Amathia convolute[J].Tetrahedron Lett, 1995, 36(16): 2783-2922.
[11] Zhang H P, Kamano Y, Ichihara Y, et al. Isolation and structure of convolutamydines B~D from marine bryozoan Amathia convolute[J].Tetrahedron ,1995, 51(19):5523-5528.
[12] Kamano Y, Kotake A, Hashima H, et al. Three New Alkaloids, Convolutamines F and G, and Convolutamydine E, from the Floridian Marine Bryozoan Amathia convolute[J].Collect Czech Chem Commun,1999, 64(7): 1147-1153.
猜你喜欢
山东第一医科大学(山东省医学科学院)学报(2021年7期)2021-10-13 06:40:42
昆明医科大学学报(2020年12期)2021-01-26 00:44:12
山东化工(2019年11期)2019-06-26 03:26:44
国际呼吸杂志(2019年1期)2019-03-08 03:07:02
铜仁学院学报(2018年6期)2018-07-05 09:47:36
中成药(2017年4期)2017-05-17 06:09:50
合成化学(2015年10期)2016-01-17 08:55:42
合成化学(2015年1期)2016-01-17 08:53:55
中国洗涤用品工业(2015年9期)2015-02-28 19:03:05
郑州大学学报(理学版)(2014年4期)2014-03-01 04:21:21
