
微波辅助高效合成N,O供体型杂环化合物
2014-08-13 01:33:36袁泽利余光勤罗玉兰宋文婷
遵义医科大学学报 2014年4期
袁泽利,余光勤,罗玉兰,宋文婷
(遵义医学院 药学院, 贵州 遵义 563099)
Macrocyclic compounds have attracted increasing interest owing to their role in the understanding of molecular processes occurring in biochemistry, pharmacy, encapsulation,activation and hydrometallurgy[1-4].
In general, several techniques such as template-assisted[5-6]or preorganization-assisted syntheses combined with the high dilution method[7]are employed for the preparation of desired macrocycles. However, synthesis of the desired macrocyclic compounds sometimes still remains problematic since competitive reactions may occur and result in several kinds of oligomeric compounds[8]. In order to circumvent those problems, people have paid more attention to the new synthetic methods. Organic reactions assisted by microwave irradiation have attracted considerable attention in recent years[9]. Additionally, a noteworthy advantage of modern scientific microwave apparatus is the ability to control reaction parameters such as temperature, pressure and reaction times accurately. So, numerous organic reactions have been performed,i.e., Michael addition, acylation and alkylation reactions,condensations, enzymatic catalysis, rearrangements, oxidations and reducetions, regioselective cycloaddition assisted by microwave irradiation[9].
In this work, we reported an efficient method for the preparation of nitrogen-oxygen donor macrocyclic compound. The synthesis of title compound (see Fig 1) was carried out more efficiently than the traditional method, using microwave irradiation in a monomode type reactor.
1 Materials and Methods
1.1 Materials All materials were, at least, of reagent grade purity and were purified or dried by standard methods before use[11].2,2′-(ethylenedioxy) bisbenzaldehyde was prepared as described previously[12].
1.2 Methods IR spectra were determined on a Varian 1000 spectrophotometer as KBr discs in the wave number range 4 000-400 cm-1. NMR spectra were recorded using a Agilent DD2 400 MHz spectrometer in CDCl3with TMS as internal standard. Microwave irradiation was performed in a commercial microwave reactor (XH-100B, 100-1000W, Beijing Xianghu Science and Technology Development Co. Ltd, Beijing, P. R. China).
1.3 Synthesis The scheme of synthesis is shown in Fig1.
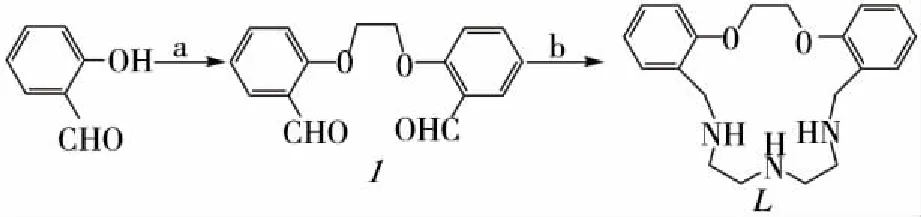
a:BrCH2CH2Br, DMF, K2CO3, 400W, 30 min, 89.2%;b:EtOH, NaBH4, 60 ℃, 400 W, 30 min, 77.7%.Fig 1 Reagents and conditions
1.3.1 General procedure for preparation of macrocyclic compound (L) The macrocyclic compound was prepared by the dropwise addition of a solution of diethylenetriamine (5 mmol) in methanol (20 ml) to a stirred solution of 1 (5 mmol) and two drops of acetic acid (catalyst) in butyl alcohol (50 ml). The reaction mixture was heated under reflux for 24 h. The solvent was removed toca. 5 ml and diethyl ether added to form yellow precipitate. The precipitate was filtered off, washed with methanol, dried, and recrystallized from methanol to give yellow compound. Then, 5 mmol of the compound was added in 20 ml methanol and reduced by the 2.5 equiv. of sodium borohydride. The colorless solution was dried under reduced pressure to afford a white mass. The mass was dissolved in 25 ml water and neutralized with dilute acetic acid. The organic product was then extracted from the aqueous solution using CH2Cl2(3×10 ml portions); removal of the solvent under reduced pressure to produce white mass, which was recrystallized from methanol to give white crystals of compound, total yield: 42.9%.
1.3.2 Microwave-assisted method for the preparation of compounds (L) In a 50 ml round-bottom flask, 1 (5 mmol) and diethylenetriamine (5 mmol) in MeOH (5 ml) were irradiated of 400 w for 30 min. Then NaBH4(11 mmol) was added and the mixture was irradiated at 400 W for 30 min. Basic workup using a saturated aqueous sodium carbonate solution and subsequent extraction using CH2Cl2was performed. The organic layer was dried over MgSO4and evaporated under reduced pressure. The crude product was washed with ethanol, dried, and recrystallized from methanol to give white crystals of compound , yield:77.7%.1HNMR spectrumin CDC13: 1.98 (s, 3H,NH),2.49-2.63 (m,8H,NCH2CH2N), 3.77 (s, 4H,arom-CH2), 4.37 (s,4H,OCH2), 6.81-7.21 ppm.(m,8H,C6H4).13C NMR(proton decoupled)spectrum in CDCl3: 48.36, 48.74(NCH2CH2N), 50.47 (arom-CH2), 66.74 (OCH2), 110.64,120.34, 128.13, 128.41, 130.78,156.98 ppm. (C6H4).FT-IR(KBr):3446,3152,1496,1454,1400,1121,1108,756.
2 Results and Discussion
2.1 Structural characteristics The characteristic spectral data are given in the experimental section. The IR spectra of title compound showed absorption bands at 3469 and 3323 cm-1due to the NH group. There were no strong bands at 1640~1600 cm-1assigned to the lack absorption of C=N. The1H NMR spectra of title compound showed a singlet at 3.77 and 4.37 ppm attributed to the arom-CH2and OCH2,at the 1.98 ppm can be assigned to protons of -NH-. However, There are multiplets at 2.49-2.63 ppm will be assigned to protons of -NCH2CH2N-. All the aromatic protons were observed in 6.81-7.21 ppm as multiplets. The corresponding13C absorption peak can also be found from13C NMR spectra, as expected. Based on the foregoing spectral data, the target compounds were assigned the cyclic structure .
2.2 The comparison of microwave irradiation and conventional heating Compared to conventional method, microwave-assisted method greatly decreased the reaction time from 24 h to 1 h and the yields were increased from 42.9% to 77.7%. The microwave-assited procedure is faster and operation simpler than ordinary thermal conditions.
3 Conclusions
In summary, we report a rapid, simple and efficient method under microwaveassisted conditions for the synthesis of nitrogen-oxygen donor macrocyclic compound. This procedure offers several advantages such as good yields of cyclization products without the need for high dilution or template conditions, shorter reaction times, and simpler operation.
[References]
[1] Lippman M, Halterman R, Perry S, et al.Glucocorticoid Binding Proteins in Human Leukaemic Lymphoblasts[J]. Nat New Biol,1973, 242(118):157-158.
[2] Reis M, Ferreira C J, Santos M M, et al. Enhancing macrocyclic diterpenes as multidrug-resistance reversers: structure-activity studies on jolkinol D derivatives[J]. J Med Chem, 2013, 56 (3): 748-760.
[3] McCauley J A, McIntyre C J, Rudd M T, et al. Discovery of vaniprevir (MK-7009), a macrocyclic hepatitis C virus NS3/4a protease inhibitor[J]. J Med Chem, 2010, 53 (6): 2443-2463.
[4] Wu Qing,Yuan Z L,Xu Y F,et al. Synthesis of New Bis-β-diketonato complexes of Titanium(IV)containing-S-heterocyclic[J]. J Zunyi Med Univ, 2010,33(3):426-430.
[5] Yuan Z L,Zhang Q L,Liang X,et al. A new series of dinucleating macrocyclic ligands and their complexes of zinc(II)[J]. Polyhedron ,2008, 27(1): 344-348
[6] Liu Y Y,Liu J,Yang J, et al. Eight coordination compounds based on a reduced Schiff base tetraaminodiphenol macrocyclic ligand [J]. Inorg Chim Acta, 2013, 403(1): 85-96.
[7] Houjou H, Lee S K, Hishikawa, et al. Highly selective formation of 2:2 macrocycles from a novel hydroxybenzaldehyde derivative and diamines[J].Chem Commun, 2000, 22(9):2197-2198.
[8] Shimakoshi H, Kai T, Aritome I,et al. Syntheses of large-membered macrocycles having multiple hydrogen bonding moieties[J].Tetrahedron Lett, 2002, 43(6):8261-8268.
[9] Surati M A, Jauhari S, Desai K R A. Purification and characterization of a novel fibrinolytic enzyme by candida guilliermondii grown on sunflower oil cake [J]. Appl Sci Res,2012, 4(2): 645-661.
[10] Greene A K, Scott L T R.Microwave-assisten perdeuteration of polycyclic aromatic hydrocarbons[J]. J Org Chem, 2013, 78:2139-2143.
[11] Dong W K,Yang R D,Yan L. Catena-poly[[(2,3-benzo-1,4,7,10,13-pentao-xa-cyclo-entadec-2-ene-κ5O) potassium]-μ-(picrato-O1,O2,O5)] [J]. Acta Crys,1998,54:1416 - 1418.
[12] Dong W K, Yang R D , Yan L. Synthesis,characterization and fluorescence of cryptates between Sm(III),Eu(III),Tb(III) and Dy(III) nitrates and a hexabenzo- cryptand[J ] . Synth React Inorg Met - Org Chem, 1999, 29 (9):1631-1640.
猜你喜欢
高等理科教育(2022年2期)2022-05-09 07:34:50
世界农药(2019年4期)2019-12-30 06:25:08
南风(2018年2期)2018-02-23 19:33:34
南风(2018年4期)2018-02-05 09:19:27
商周刊(2017年10期)2017-08-23 13:30:40
学生天地(2017年10期)2017-05-17 05:50:28
合成化学(2015年2期)2016-01-17 09:03:25
合成化学(2015年9期)2016-01-17 08:57:21
无机化学学报(2014年1期)2014-02-28 17:30:01
天然产物研究与开发(2012年4期)2012-12-22 09:01:16
