
通过测量模拟样品中236U与238U原子个数比估算燃耗的探索研究
2014-08-08 08:24:24姜小燕常志远赵永刚张继龙李力力
原子能科学技术 2014年8期
姜小燕,常志远,赵永刚,张继龙,李力力
(中国原子能科学研究院 放射化学研究所,北京 102413)
核事故造成的人工长寿命放射性同位素环境污染成为近几十年来最严重的环境问题之一[1-2]。环境监测及污染源(核武器试验、核电厂以及核燃料后处理厂等)的评估需精确分析锕系元素的同位素,尤其是铀、钚同位素[3]。后处理厂周围受污染环境中的铀、钚同位素组成可指示乏燃料燃耗,通过燃耗可推断铀燃料的利用率、裂变产物及人工锕类元素的生成量[4-5]。
人类核活动(如乏燃料的处理)所带入环境中的铀远低于环境中的天然铀含量,但其同位素组成与天然铀同位素却有很大差别,通过对被污染土壤中铀同位素及含量的分析可推断附近核设施中反应堆燃料铀的富集度、燃耗和以往的核事故信息[5],在环境保护、核安全和核保障核查领域具有重要意义。
文献[1]分析了不同类型的ICP-MS结合不同的进样装置测量236U/238U的实验条件及其测量结果,发现ICP-MS测量236U的灵敏度较Alpha谱仪的好两个量级[2]。文献[4]对切尔诺贝利核电站受污染区域的土壤取样,用ICP-MS分析235U/238U(235U与238U原子个数比)、236U/238U(236U与238U原子个数比),得出堆后铀燃耗等级的微小变化。本工作利用MC-ICP-MS多接收器测量同位素比高精度的优点,测量模拟样品中236U/238U并估算模拟样品的燃耗。
1 原理
自然界中236U的含量极低(236U/238U约10-14),近似可认为自然界中不存在236U。在乏燃料处理过程中,堆后铀(含一定量的236U)可能通过气、液流出物等形式排放释入环境,并与环境中的天然铀混合。通过分析环境样品中的235U/238U、236U/238U可简单判断堆后铀燃耗。假定环境样品中铀的来源为堆后铀与天然铀,则其中的235U/238U、236U/238U与燃耗的关系用式(1)、(2)表示[3]。
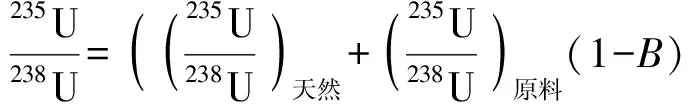
(1)
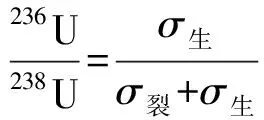
(2)
式中:p为受污染土壤中的乏燃料铀与天然铀总量的比值;B为燃耗;σ生、σ裂分别为235U生成236U和235U裂变的中子吸收截面。
假定天然铀丰度、反应堆燃料铀的初始富集度已知,环境样品中的同位素比可实验测量,则式(1)、(2)中的B、p可通过迭代的方法计算得出,也可利用式(1)、(2)通过求解B、p的计算程序得到。
环境样品中的235U/238U、236U/238U用MC-ICP-MS测量,MC-ICP-MS的多接收器测量同位素比精度高,其Daly接收器用来接收低含量的236U,不但提高了测量灵敏度且降低了大量238U对236U测量的干扰。
2 实验
2.1 仪器和试剂
Isoprobe型MC-ICP-MS(英国GV);APEX高效进样系统(美国Elemental Scientific);Aridus高效进样系统(美国CETAC);PFA普通雾化进样系统(美国Savillex)。
CRM 0002、CRM 005A铀同位素标准物质(NBL);UTB010铀同位素工作标准物质(中国原子能科学研究院)。
2.2 MC-ICP-MS测量铀同位素比的优化条件实验
由于236U的含量很少,且235U与238U的大量存在干扰236U的测量,为精确测定236U,需用超灵敏的同位素分析技术。MC-ICP-MS配置六极杆碰撞池及多接收器,可降低235UH+对236U+的干扰并提高测量精度。本研究分别采用Aridus、APEX高效微量进样器及PFA普通雾化进样器进样,利用MC-ICP-MS对236U/238U的测量方法进行研究。表1、2分别为仪器的测量参数及接收器的排布,特别采用Daly接收器测量CRM U0002标准溶液中的238UH+,以观察铀氢峰的形成情况。
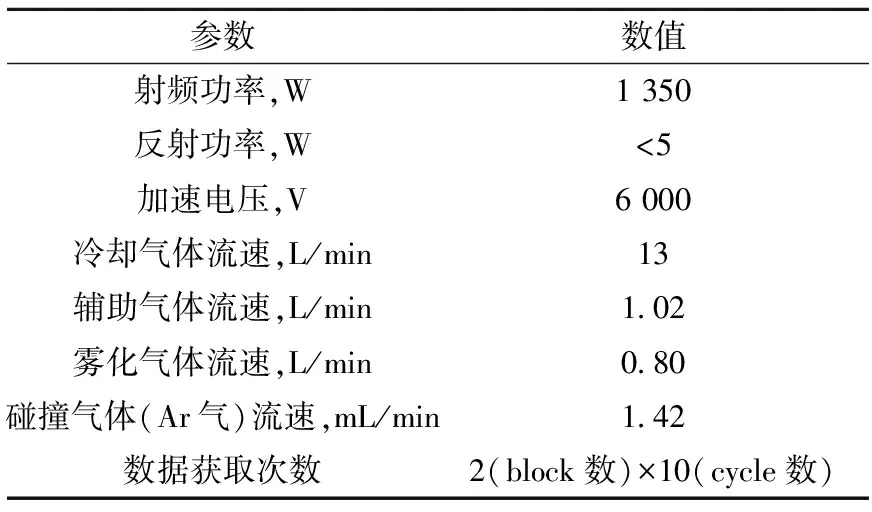
表1 MC-ICP-MS测量的参数

表2 MC-ICP-MS接收器的排布
2.3 模拟样品配制
将天然铀与堆后铀样品按不同比例混合,配备出一系列燃耗固定而堆后铀与天然铀之比不同的样品。
实验时,堆后铀样品是235U丰度为3.0%的铀样品经两次不同条件的辐照产生的,分别简称为照后1、照后2。将照后1、照后2与天然铀按不同比例混合,得到两批模拟样品(混合样1、混合样2)。配比列于表3。

表3 模拟样品配比
3 结果与讨论
3.1 MC-ICP-MS测量236U/238U
1) 多原子离子的干扰
表4 多原子离子的计数率
2)235UH+峰的干扰
测量铀贫化度较高的CRM 0002标准溶液中的238UH+/238U+,以此类推235UH+对236U+的贡献, Aridus、APEX及普通进样系统下235UH+峰的生成量如图1所示。
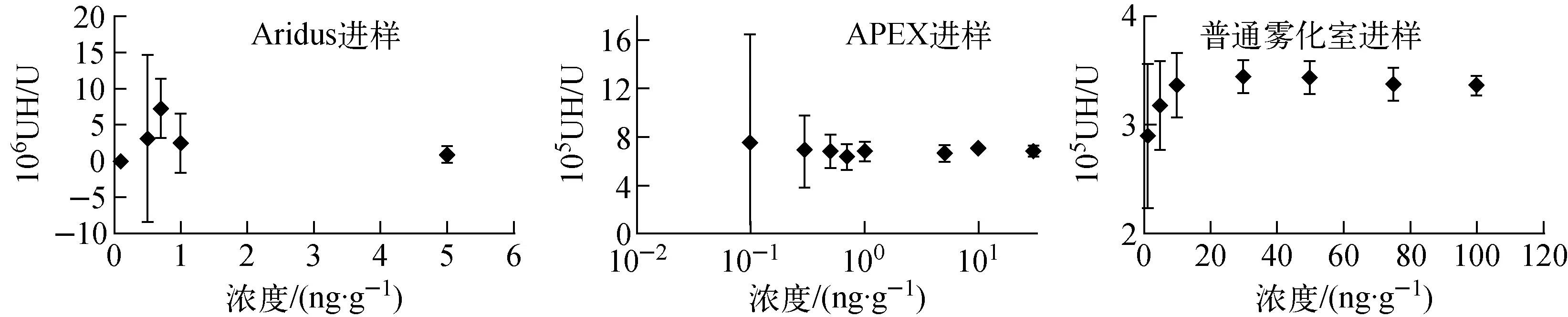
图1 不同进样系统下的235UH+生成量
采用APEX和普通进样器进样时,UH+/U均为10-5量级,Aridus独有的膜去溶功能抑制了UH+的生成,在Aridus进样条件下UH+/U降为10-6量级。无论采取何种进样方式,随样品浓度的变化,UH+/U出现不同程度的波动,扣除235UH+干扰时,需根据不同的样品浓度分析235UH+对236U+的贡献。
将CRM 005A标准物质作为待测样品,UTB010作为标准物质,采用k校正法(线性校正法)修正测量过程中的质量歧视效应,测量结果及扩展不确定度列于表5。
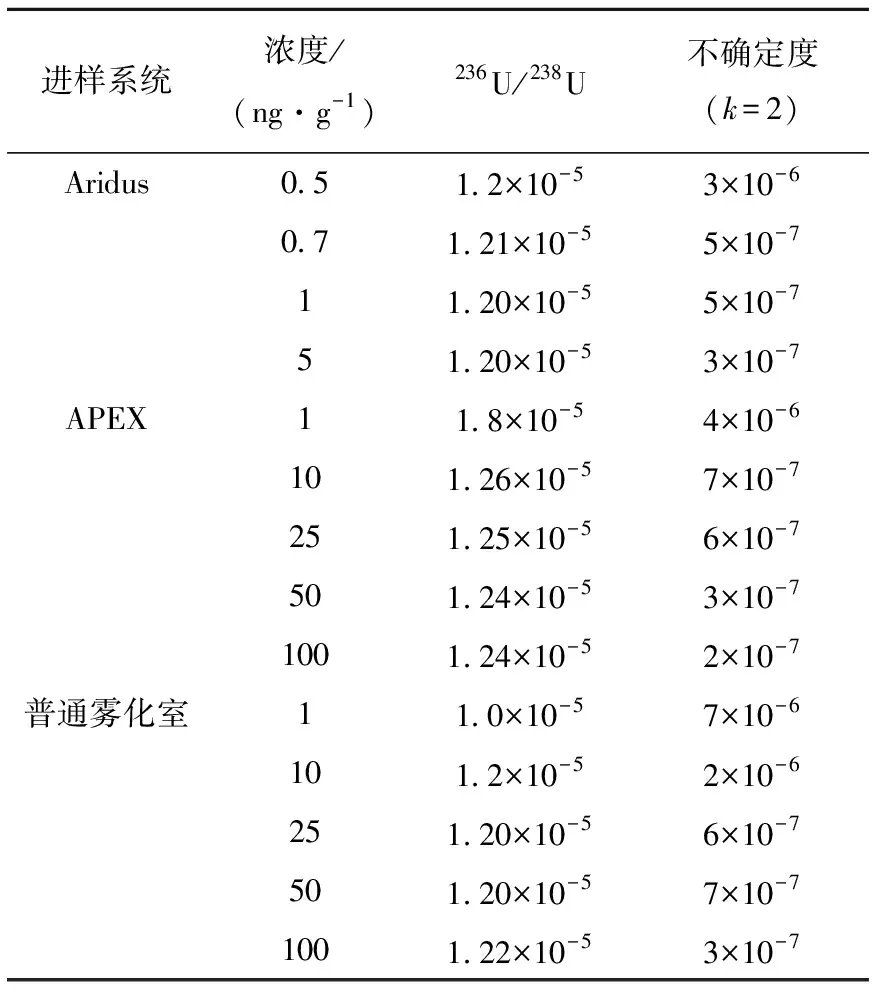
表5 3种进样系统下CRM 005A的测量结果及其不确定度
采用Aridus进样,对浓度小于0.5 ng/g的CRM 005A溶液,其中的236U含量很小,测量结果相对不确定度大于10%,且偏离参考值;当铀浓度达1 ng/g,即236U浓度约达0.01 pg/g时,测量结果的相对不确定度小于5%,测量值与参考值(1.19±0.01)×10-5在不确定度范围内一致,且较稳定。
3.2 模拟环境样品中235U/238U、236U/238U的测量及燃耗估算
1)235U/238U、236U/238U的测量结果
表6、7为模拟混合样品的235U/238U、236U/238U测量结果,扩展不确定度合成时考虑了样品测量、标准物质测量、标准物质标称值的不确定度。

表6 混合样1的测量结果
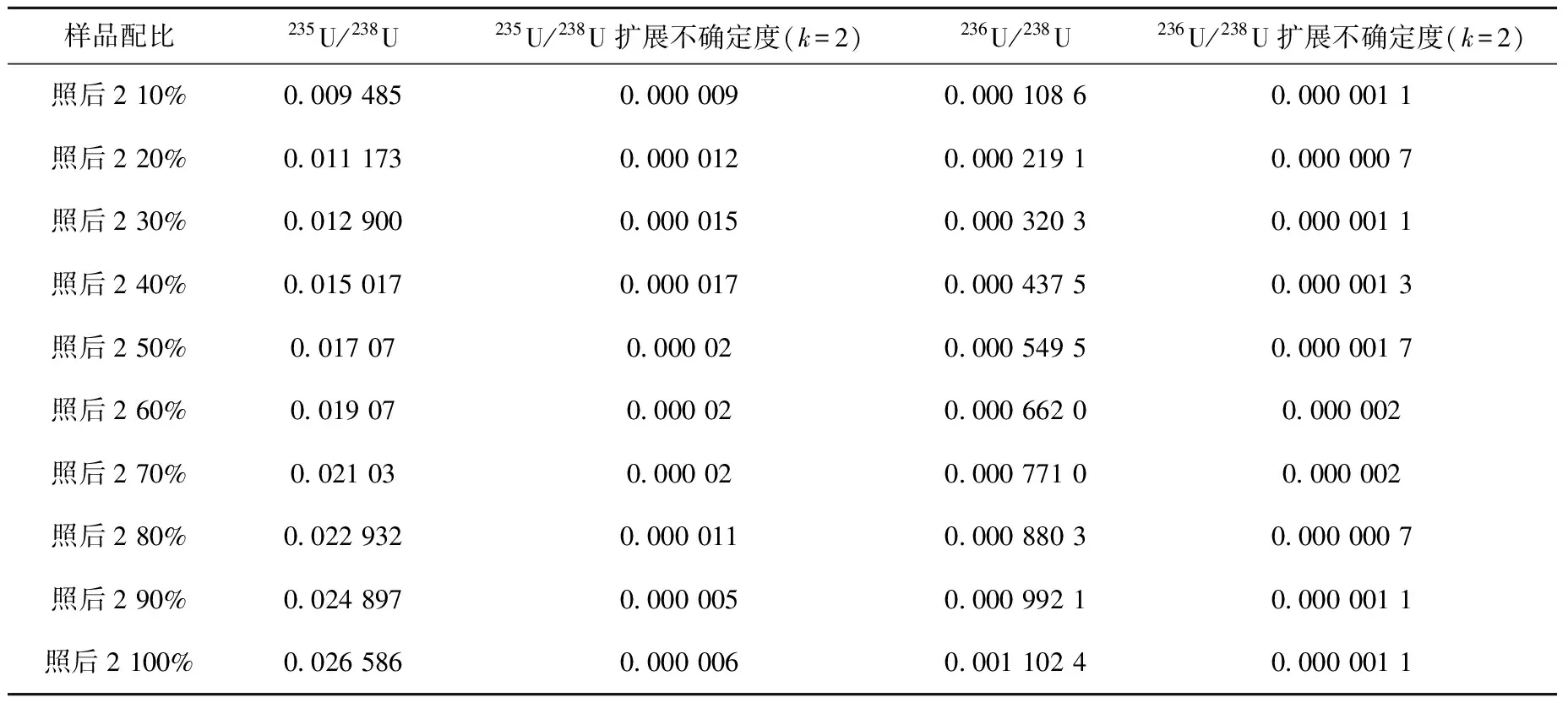
表7 混合样2的测量结果
2) 模拟样品中B的计算及与理论值的比较
用文献[3]中的235U/238U、236U/238U数据对编制的燃耗计算程序进行了验证,结果列于表8。结果表明,文献值与计算值基本一致,相对偏差小于2.5%,说明计算程序可用于燃耗推算。
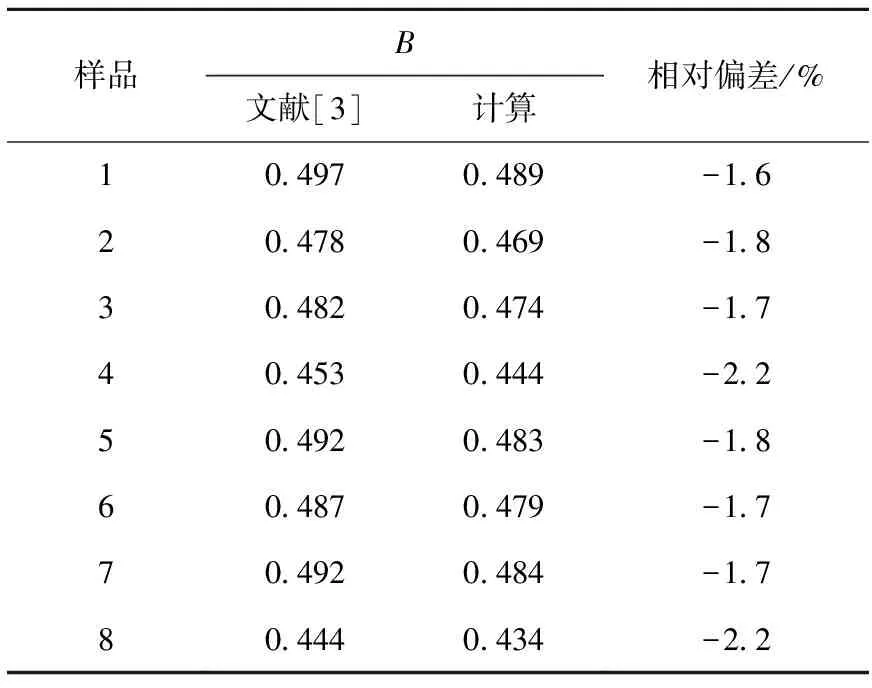
表8 燃耗计算结果验证
对本实验测得的模拟混合样品的235U/238U和236U/238U(表6、7数据),利用编制的计算程序进行燃耗计算,结果列于表9。两种燃耗的样品与不同比例的天然铀混合后,通过铀同位素测量和程序计算得到的燃耗在不同比例时的结果基本一致。
对两个原始辐照样采用ORIGEN2燃耗计算程序和式(3)也进行了计算。
(3)
式中:(236U/235U)1、(236U/235U)0分别为辐照后和初始燃料中的236U/235U;α5为235U的中子俘获与裂变截面之比。
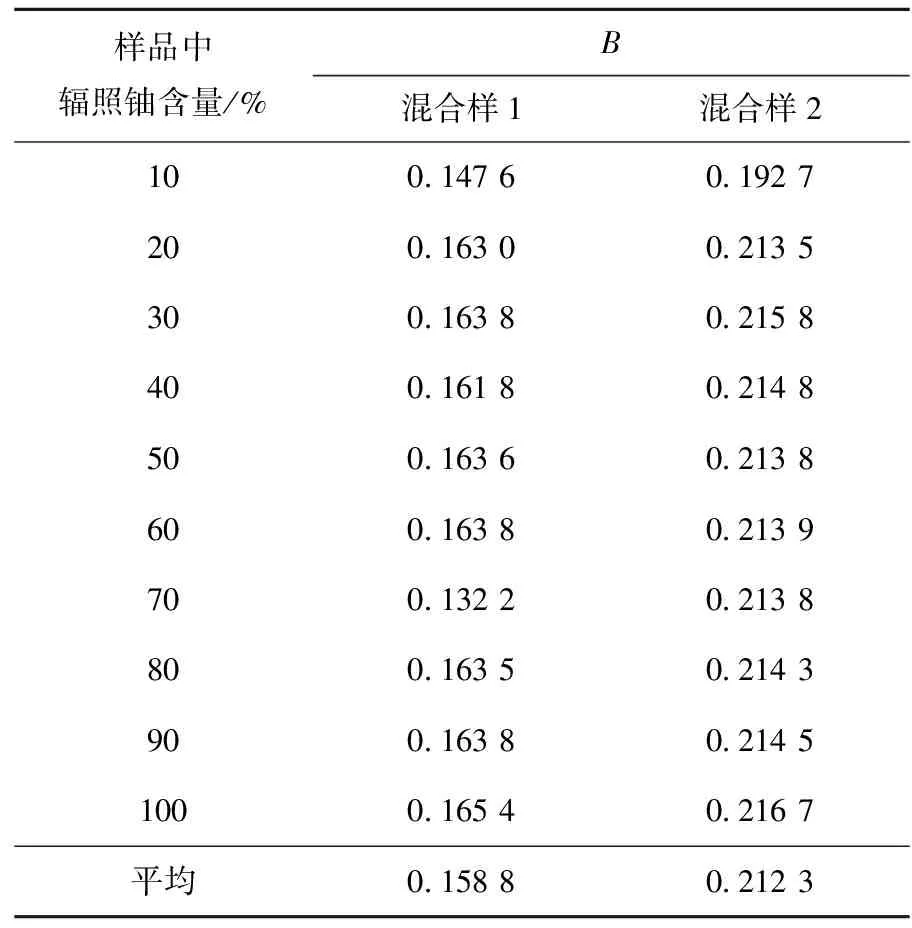
表9 模拟样品的燃耗计算结果
ORIGEN2程序是利用初始燃料中的铀同位素含量、辐照的堆型、辐照时间、中子通量等参数计算燃耗;式(3)是通过辐照前、后燃料中的236U/235U的变化来计算燃耗;而本工作是通过分析受堆后铀污染的环境样品中236U/238U、235U/238U,结合辐照前燃料的初始235U/238U推出堆后铀的燃耗,无需了解反应堆类型及辐照参数,分析环境样品即可推知燃耗,难度相对较大。燃耗计算结果的比较列于表10。
从表10可知,ORIGEN2、编制程序、公式计算的燃耗结果总体较接近,但也存在着10%左右的相对偏差。出现以上差别的原因分析如下:ORIGEN2在实际计算燃耗时,仅考虑了辐照时间,且认为热中子、中子通量不变,未考虑开停堆的变化;编制程序所采用的式(1)~(3)本身为近似公式,忽略了部分因素的近似结果,且计算时虽考虑了开停堆所导致的中子通量的变化,但却认为所有的中子均为热中子,以上因素均可能导致不同方法之间计算结果的偏差。
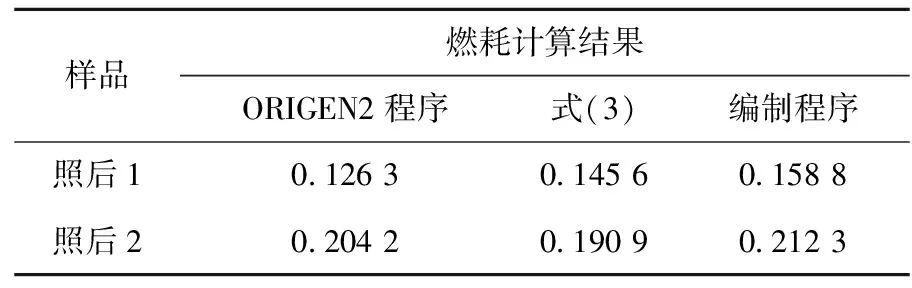
表10 燃耗计算结果比较
4 结论
本工作在优化MC-ICP-MS测量条件的基础上,建立了MC-ICP-MS精密测定236U/238U的方法,研究了测定过程中Pb及235UH+的影响。结果表明:采用Aridus系统进样时,Pb含量小于10 ng·g-1时不会对236U测量产生影响;UH的影响约为10-6。
采用质谱测定235U/238U和236U/238U及自编程序,对混有不同比例天然铀的辐照铀样品燃耗进行了估算,估算值与采用其他方法得到的燃耗较接近,相对偏差约为10%。对于单一污染源的环境样品,可通过235U/238U和236U/238U粗略推算污染源的燃耗。
参考文献:
[1] BOULYGA S F, MATUSEVICH J L, MIRONOV V P, et al. Determination of236U/238U isotope ratio in contaminated environmental samples using different ICP-MS instruments[J]. J Anal At Spectrom, 2002, 17: 958-964.
[2] BOULYGA S F, HEUMANN K G. Determination of extremely low236U/238U isotope ratios in environmental samples by sector-field inductively coupled plasma mass spectrometry using high-efficiency sample introduction[J]. Journal of Environmental Radioactivity, 2006, 88: 1-10.
[3] BOULYGA S F, BECKER J S. Isotopic analysis of uranium and plutonium using ICP-MS and estimation of burn-up of spent uranium in contaminated environment samples[J]. J Anal At Spectrom, 2002, 17: 1 143-1 147.
[4] MIRONOV V P, MATUSEVICH J L, KUDRJASHOV V P, et al. Determination of uranium concentration and burn-up of irradiated reactor fuel in contaminated areas in Belarus using uranium isotopic ratios in soil samples[J]. Radiochim Acta, 2005, 93: 781-784.
[5] BOULYGA S F, BECKER J S. Determination of uranium isotopic composition and236U content of soil samples and hot particles using inductively coupled plasma mass spectrometry[J]. Fresenius J Anal Chem, 2001, 370: 612-617.
猜你喜欢
甘肃科技(2020年20期)2020-04-13 00:30:40
核技术(2016年4期)2016-08-22 09:05:28
核科学与工程(2016年3期)2016-01-03 07:22:52
电源技术(2015年7期)2015-08-22 08:49:04
同位素(2014年3期)2014-06-13 08:22:28
核科学与工程(2014年3期)2014-05-11 02:57:18
同位素(2014年2期)2014-04-16 04:57:15
同位素(2014年2期)2014-04-16 04:57:12
火炸药学报(2014年3期)2014-03-20 13:17:39
电力自动化设备(2013年11期)2013-09-18 02:55:06
