
奋乃静在人胆汁中的代谢物分析
2014-08-07 02:22尤天庚杜江波陈笑艳钟大放
质谱学报 2014年6期
尤天庚,谢 岑,杜江波,陈笑艳,钟大放
(1. 同济大学附属东方医院普外科,上海 200120;2. 中国科学院上海药物研究所,上海 201203)
奋乃静(perphenazine)为吩噻嗪类多巴胺D2受体阻断剂,临床上主要用于治疗精神分裂症、躁狂症,也可用于治疗呕吐及严重焦虑。Symchowicz等[1]研究了35S-奋乃静在大鼠体内的分布和排泄。皮下注射0.3 mg/kg的35S-奋乃静后,药物主要分布在肺、肝、肾、脾和垂体中,24 h内超过80%的药物被排泄,其中的80%经粪便排泄,这表明奋乃静在大鼠体内主要经胆汁排泄。Eggert等[2]报道了奋乃静在人体内的药动学。静脉注射5 mg奋乃静后,体内有重吸收现象,半衰期约9.5 h,提示奋乃静在人体内可能发生肝肠循环。Huang等[3]报道了奋乃静在精神分裂症患者和健康人体内的代谢和排泄。尿中主要代谢途径包括S-氧化、吩噻嗪环上7位羟基化和葡萄糖醛酸结合等;在给药后17周内,尿中仅回收了给药剂量的44%,表明奋乃静在人体内除尿排泄外,尚存在主要排泄途径未被阐明。因此,为全面地了解奋乃静在人体内的代谢过程,研究其胆汁排泄十分必要。
胆汁排泄能够显著影响某些药物的系统暴露量、药理作用和毒性。由于人胆汁样品不易获得,通常使用受试者粪样替代,用以研究药物的胆汁排泄。但是这种方法存在着诸多弊端:首先,该方法无法区分胆汁排泄和肠道外排过程,对于口服给药的药物,该方法同样不能区分未吸收的药物和吸收后经胆汁排泄或者肠道外排的药物,因此粪样中药物及代谢物的剂量和相对比例并不能真正反映胆汁排泄的情况。另外,一些不稳定的代谢物(如葡萄糖醛酸结合物)可能在肠菌群的作用下发生降解后排出体外,或者通过肠肝循环再重吸收进入人体循环,这些过程都将导致粪样无法准确地反映药物的胆汁排泄过程[4-7]。因此,直接研究人胆汁中的代谢物,可以更可靠地确定药物及其代谢物在肝脏、胆囊和胆道中的暴露情况[8-18]。
胆汁是一种复杂的生物基质,主要组成是胆酸类化合物,相对分子质量约400 Da,与大多数药物的相对分子质量接近,其种类和结构的多样性给胆汁中药物代谢物的鉴定带来了巨大挑战。UPLC/Q-TOF MS结合了MDF[19]和generic dealkylation技术[20],能有效去除内源性物质的干扰,MSE利用碰撞能量梯度,可同时完成对前体离子和产物离子的采集,显著缩短了分析时间[21]。本研究利用T管引流技术收集患者服用奋乃静片后的胆汁,采用UPLC/Q-TOF MS法快速表征奋乃静在胆汁中的代谢物,以进一步完善奋乃静在人体内的代谢途径。
1 实验部分
1.1 仪器与装置
Acquity Ultra超高效液相色谱系统及Synapt Q-TOF质谱仪:美国Waters公司产品, 配有Masslynx V4.1,MetaboLynx及MassFragmentTM数据处理系统。
1.2 材料与试剂
奋乃静片:由上海市东方医院提供;乙腈、甲酸(色谱纯):德国Fluka公司产品;醋酸铵(色谱纯):美国Tedia公司产品;β-葡萄糖苷酸酶:美国Sigma公司产品;枸橼酸钠:国药集团化学试剂有限公司产品;去离子水:由法国Millipore纯水仪制备。
1.3 胆汁样品采集
对一名患有精神分裂症的胆管结石患者实施胆管插管手术后,中、晚各服用4 mg奋乃静,收集首次给药后24 h内的胆汁样品,于-20 ℃保存待测。本实验按照国家食品药品监督管理局GCP指导原则设计方案,并获得上海东方医院伦理委员会批准,实验前受试者自愿签署书面知情同意书。
1.4 分析方法
1.4.1 色谱条件 Acquity UPLC HSS T3色谱柱(2.1 mm×100 mm×1.8 μm);柱温40 ℃;流动相: A(5 mmol/L醋酸铵水溶液),B(0.05%甲酸-乙腈溶液);流速0.4 mL/min;梯度程序洗脱:0~3 min,5% B,3~16 min,5%~45% B,16~18 min,45% B,18~20 min,45%~100% B,20~25 min,100%~5% B。
1.4.2 质谱条件 ESI离子源,正离子方式检测,雾化气(氮气)流量700 L/h,去溶剂温度350 ℃,离子源温度120 ℃,毛细管电压3.0 kV。低能量扫描时,传输碰撞能量3 eV,阱碰撞能量5 eV;高能量扫描时,传输碰撞能量15 eV,阱碰撞能量15~35 eV。质量扫描范围m/z80~1 000,质谱分辨率约为10 000。选取400 μg/L亮氨酸-脑啡肽(m/z556.277 1)作为质荷比的外标校正(Lock SprayTM,5 μL/min)。
1.5 样品预处理
向300 μL胆汁样品中加入600 μL乙腈,涡流混合1 min,以11 000 r/min离心5 min,取出全部上清液置于10 mL试管中,40 ℃氮气流下吹干,残留物以100 μLV(5 mmol/L醋酸铵水溶液)∶V(0.05%甲酸-乙腈溶液)=9∶1的溶液溶解,取10 μL进行UPLC/Q-TOF MS分析。
向300 μL胆汁样品中加入200 μL 2 000 U/mL的β-葡萄糖苷酸酶(由pH 5枸橼酸钠缓冲液配制),37 ℃下水浴温孵16 h后,迅速冷却,按1.5预处理后,进行UPLC/Q-TOF MS分析。
2 结果与讨论
2.1 奋乃静对照品色谱和质谱分析
对奋乃静对照品溶液进行UPLC/Q-TOF MS分析,利用MSE功能获得低、高碰撞能量下的质谱信息:低碰撞能量下获得前体离子质谱图,高碰撞能量下获得产物离子信息。奋乃静的色谱保留时间为15.33 min,由于其分子中含有1个氯原子,也可以通过同位素峰簇辅助解析其裂解途径。
在低碰撞能量质谱图中,奋乃静的[M+H]+离子为m/z404/406(C21H27ClN3OS+)。在高碰撞能量质谱图中,主要碎片离子为m/z274/276、246/248、171、143、100、98,示于图1。m/z171(C9H19N2O+)为前体离子经i-断裂丢失2-氯吩噻嗪(C12H8ClNS)分子后生成的碎片离子,相对丰度为100%;m/z274/276(C15H13ClNS+)及m/z246/248(C13H9ClNS+)两对离子的丰度比均为3∶1,证明碎片离子中含有氯原子,前者为前体离子经i-断裂丢失1-哌嗪乙醇(C6H14N2O)分子生成的碎片离子,后者为前体离子经氢重排后丢失4-乙基-1-哌嗪乙醇(C8H18N2O)分子后生成的碎片离子;m/z143(C7H15N2O+)为前体离子经氢重排后丢失10-乙基-2-氯吩噻嗪(C14H12ClNS)分子后生成的碎片离子;m/z100(C5H10NO+)为前体离子经i-断裂和多步氢重排后生成的碎片离子;m/z98(C5H10N2+)为前体离子经多步重排后生成的碎片离子。
根据奋乃静的裂解特点,可将其结构分为A(1-哌嗪乙醇)、B(2-氯吩噻嗪)两个片段。代谢物是原形药物在体内经酶催化产生的生物转化产物,与原形药物具有相似的母核结构,因此根据原形药物的质谱裂解规律,可以推测代谢物的结构[22]。
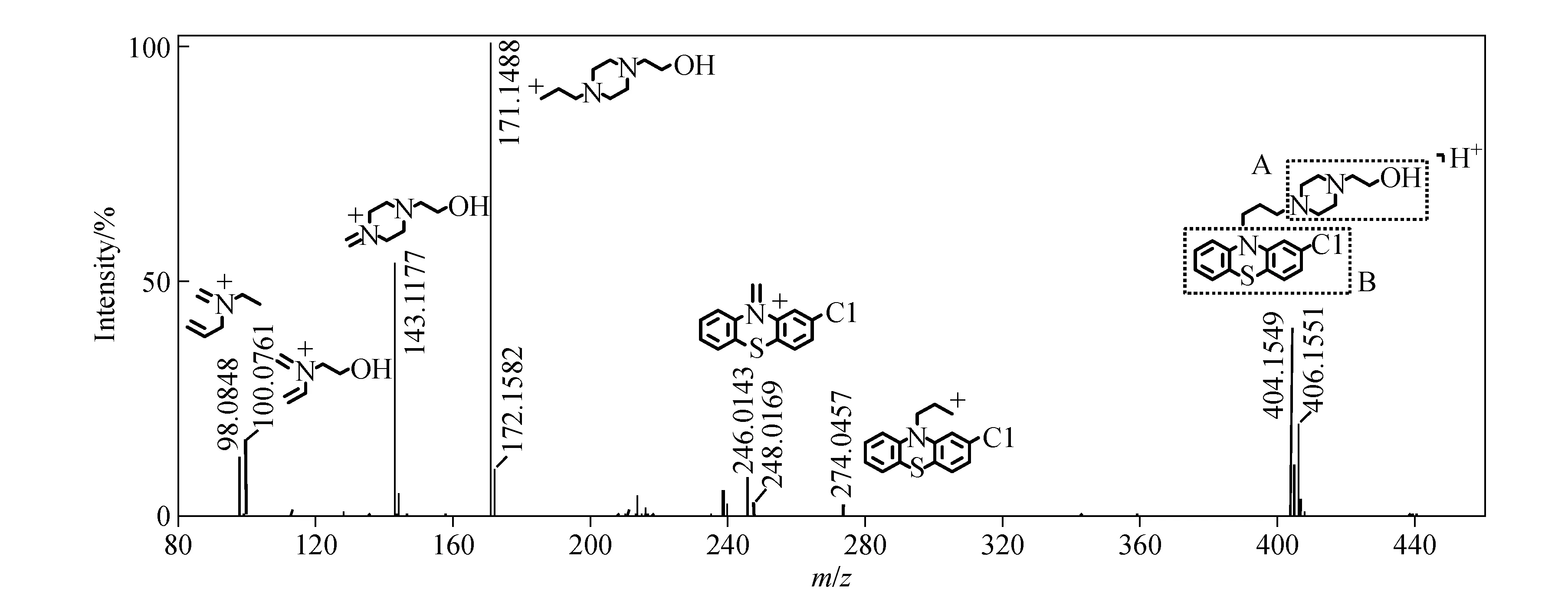
图1 奋乃静对照品在高碰撞能量下的质谱图Fig.1 Mass spectra of perphenazine under high collision energy
2.2 人胆汁中的代谢产物鉴定
采用MDF和generic dealkylation等软件对MSE数据进行处理,获得胆汁样品中代谢物的色谱图,示于图2。与空白胆汁样品的色谱图比较,在服药后的人胆汁中主要发现16对相关离子,分别为m/z404/406、376/378、392/394、394/396、418/420、420/422、434/436、436/438、500/502、530/532、552/554、580/582、596/598、610/612、612/614、626/628,每对离子的丰度比均为3∶1,分别命名为M0~M15。奋乃静代谢物的准确分子质量、可能元素组成和代谢途径等列于表1。本工作以奋乃静的主要代谢产物M5、M12和M15为例,阐述代谢物的结构鉴定过程。
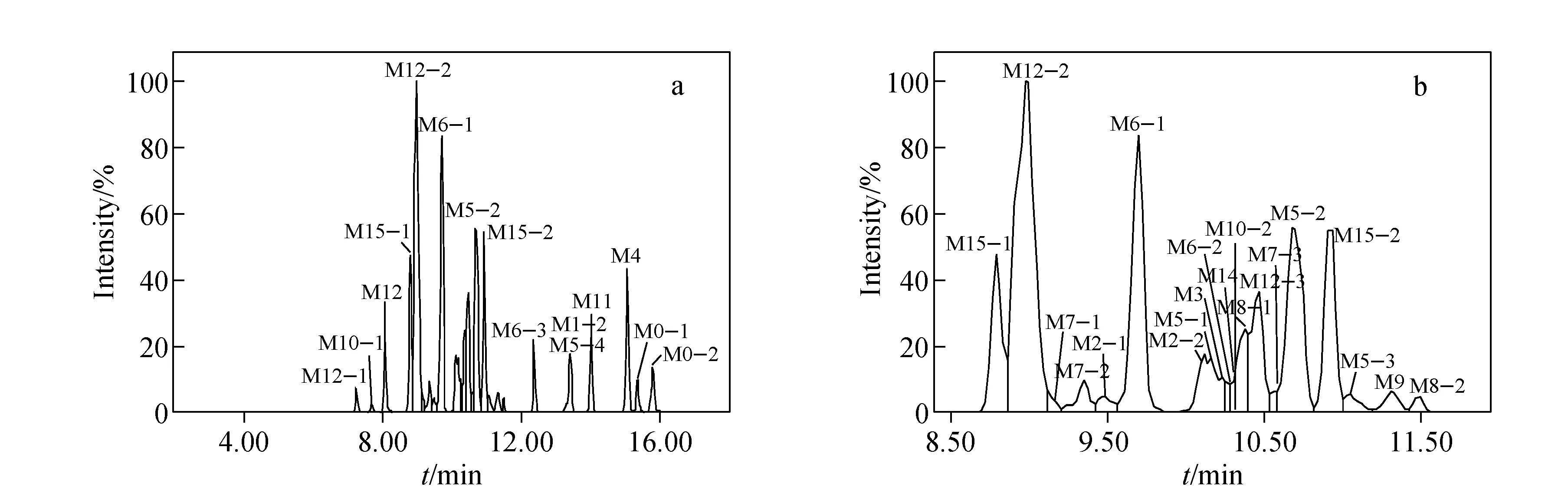
a. 全图;b. 8.5~12 min放大图图2 服药后人胆汁中奋乃静代谢物的色谱图Fig.2 Chromatograms of perphenazine metabolites in human bile
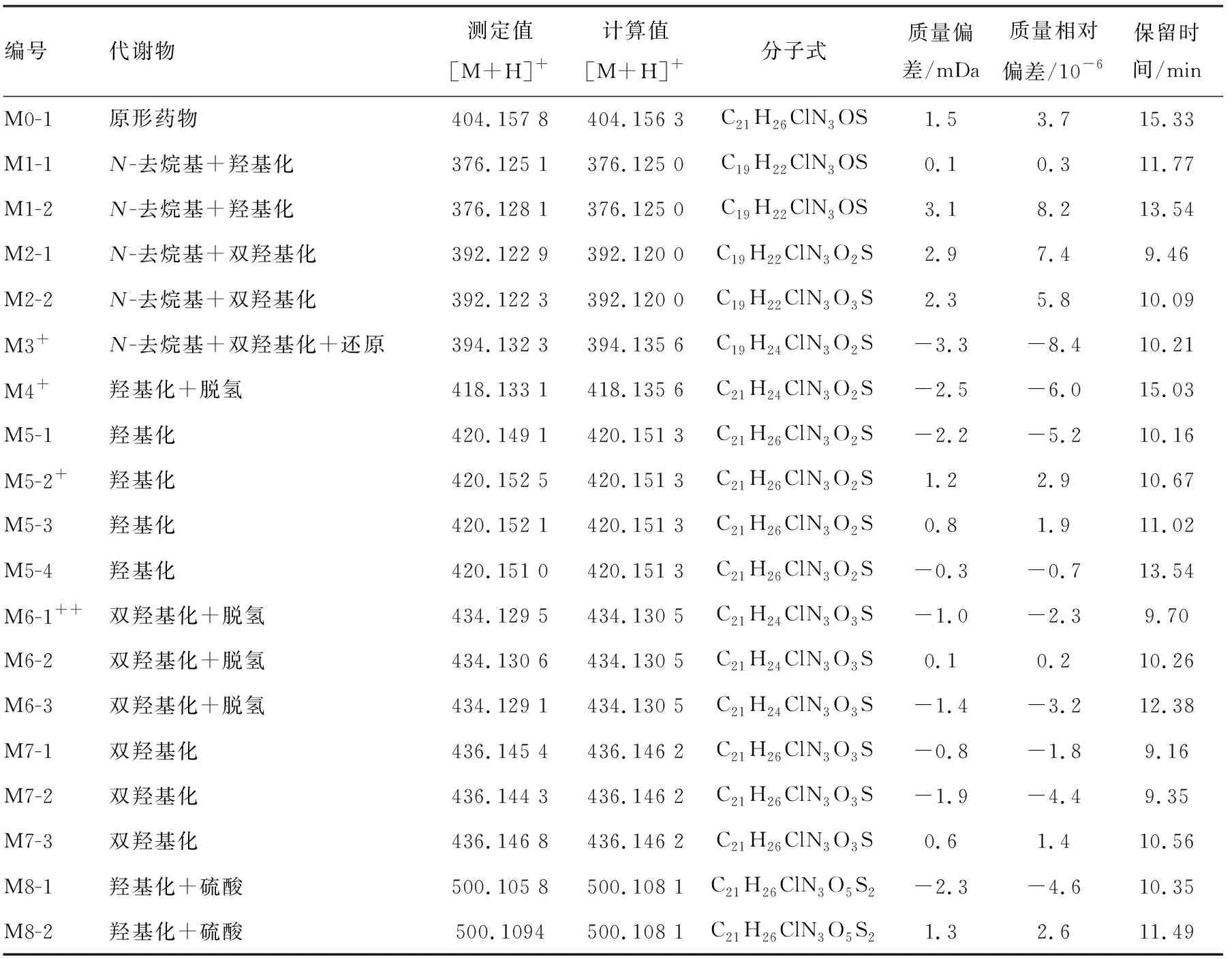
编号代谢物测定值[M+H]+计算值[M+H]+分子式质量偏差/mDa质量相对偏差/10-6保留时间/minM0-1原形药物404.157 8404.156 3C21H26ClN3OS1.53.715.33M1-1N-去烷基+羟基化376.125 1376.125 0C19H22ClN3OS0.10.311.77M1-2N-去烷基+羟基化376.128 1376.125 0C19H22ClN3OS3.18.213.54M2-1N-去烷基+双羟基化392.122 9392.120 0C19H22ClN3O2S2.97.49.46M2-2N-去烷基+双羟基化392.122 3392.120 0C19H22ClN3O3S2.35.810.09M3+N-去烷基+双羟基化+还原394.132 3394.135 6C19H24ClN3O2S-3.3-8.410.21M4+羟基化+脱氢418.133 1418.135 6C21H24ClN3O2S-2.5-6.015.03M5-1羟基化420.149 1420.151 3C21H26ClN3O2S-2.2-5.210.16M5-2+羟基化420.152 5420.151 3C21H26ClN3O2S1.22.910.67M5-3羟基化420.152 1420.151 3C21H26ClN3O2S0.81.911.02M5-4羟基化420.151 0420.151 3C21H26ClN3O2S-0.3-0.713.54M6-1++双羟基化+脱氢434.129 5434.130 5C21H24ClN3O3S-1.0-2.39.70M6-2双羟基化+脱氢434.130 6434.130 5C21H24ClN3O3S0.10.210.26M6-3双羟基化+脱氢434.129 1434.130 5C21H24ClN3O3S-1.4-3.212.38M7-1双羟基化436.145 4436.146 2C21H26ClN3O3S-0.8-1.89.16M7-2双羟基化436.144 3436.146 2C21H26ClN3O3S-1.9-4.49.35M7-3双羟基化436.146 8436.146 2C21H26ClN3O3S0.61.410.56M8-1羟基化+硫酸500.105 8500.108 1C21H26ClN3O5S2-2.3-4.610.35M8-2羟基化+硫酸500.1094500.108 1C21H26ClN3O5S21.32.611.49
续表
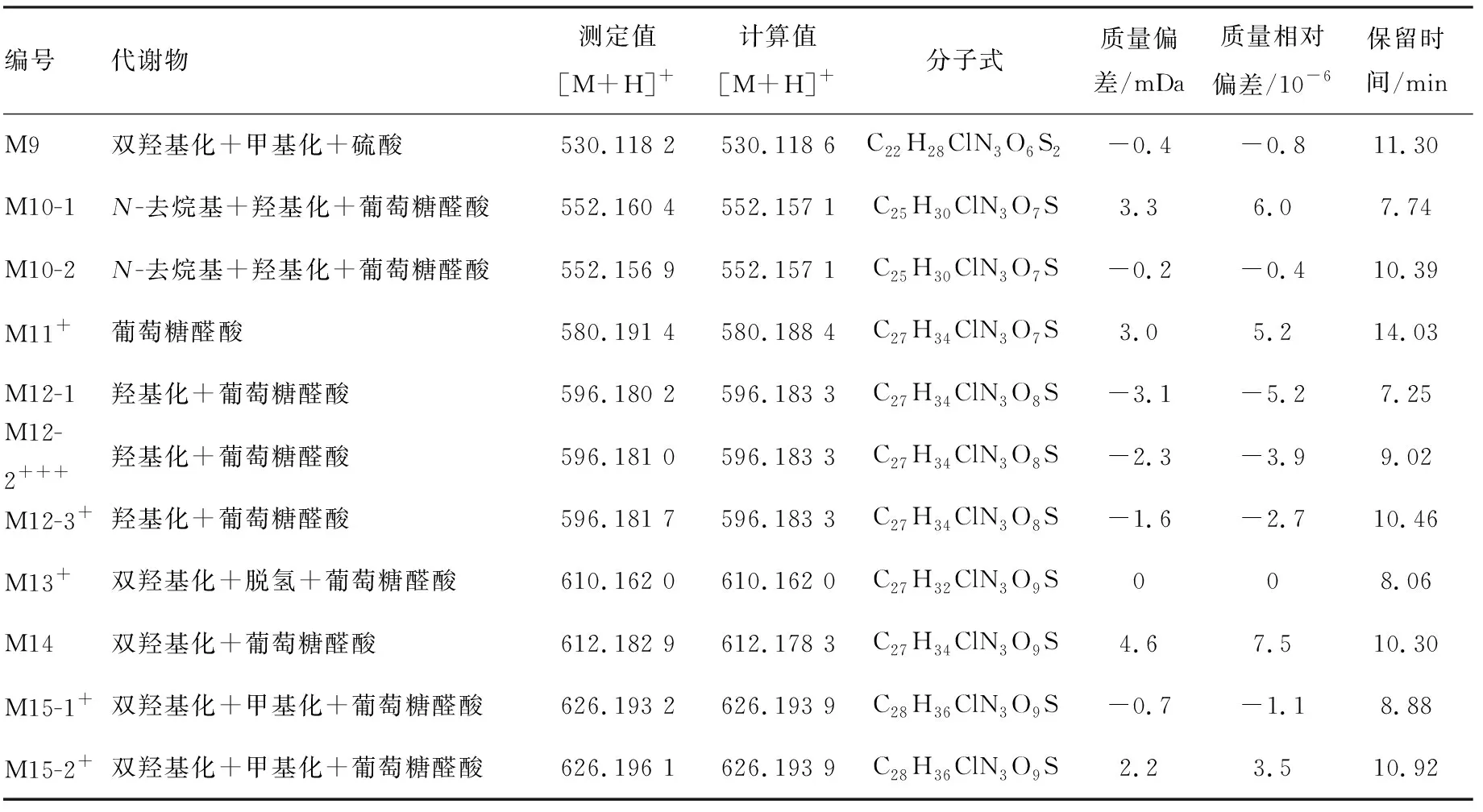
编号代谢物测定值[M+H]+计算值[M+H]+分子式质量偏差/mDa质量相对偏差/10-6保留时间/minM9双羟基化+甲基化+硫酸530.118 2530.118 6C22H28ClN3O6S2-0.4-0.811.30M10-1N-去烷基+羟基化+葡萄糖醛酸552.160 4552.157 1C25H30ClN3O7S3.36.07.74M10-2N-去烷基+羟基化+葡萄糖醛酸552.156 9552.157 1C25H30ClN3O7S-0.2-0.410.39M11+葡萄糖醛酸580.191 4580.188 4C27H34ClN3O7S3.05.214.03M12-1羟基化+葡萄糖醛酸596.180 2596.183 3C27H34ClN3O8S-3.1-5.27.25M12-2+++羟基化+葡萄糖醛酸596.181 0596.183 3C27H34ClN3O8S-2.3-3.99.02M12-3+羟基化+葡萄糖醛酸596.181 7596.183 3C27H34ClN3O8S-1.6-2.710.46M13+双羟基化+脱氢+葡萄糖醛酸610.162 0610.162 0C27H32ClN3O9S008.06M14双羟基化+葡萄糖醛酸612.182 9612.178 3C27H34ClN3O9S4.67.510.30M15-1+双羟基化+甲基化+葡萄糖醛酸626.193 2626.193 9C28H36ClN3O9S-0.7-1.18.88M15-2+双羟基化+甲基化+葡萄糖醛酸626.196 1626.193 9C28H36ClN3O9S2.23.510.92
注:+++为代谢物比例>20%;++为代谢物比例>10%;+为代谢物比例>5%;未标注的表示代谢物比例<5%
2.2.1 M0([M+H]+,m/z404/406)
从MDF色谱图中选择性检测m/z404/406,在保留时间为15.33、15.77 min出现2个色谱峰,命名为M0-1和M0-2,根据准确分子质量,推测它们的分子式均为C21H26ClN3OS,M0-1的色谱保留时间及高能量下的主要碎片离子均与奋乃静对照品相同,从而确定M0-1是未被代谢的原形药物奋乃静。M0-2的结构有待进一步考察。
2.2.2 M5([M+H]+,m/z420/422)
从MDF色谱图中选择性检测m/z420/422(m/z404/406+16 Da),在保留时间为10.16、10.67、11.02、13.54 min出现4个色谱峰,分别命名为M5-1、M5-2、M5-3和M5-4,根据准确分子质量,推测它们的分子式均为C21H26ClN3O2S(原形分子式为C21H26ClN3OS),比原形多了1个O。在高碰撞能量下,M5-1和M5-3均产生m/z274/276、246/248、187、159碎片离子,其中m/z274/276和m/z246/248的碎片离子与原形药物的B片段相同,而m/z187和m/z159的碎片离子均比原形药物的A片段多16 Da,推测M5-1和M5-3为M0的A片段羟基化代谢物。而M5-2和M5-4产生了m/z290/292、262/264、171、143、100、98碎片离子,其中m/z171、143、100、98的碎片离子与原形药物的A片段相同,而m/z290/292、262/264的碎片离子均比原形药物的B片段多16 Da,推测M5-2和M5-4为M0的B片段羟基化代谢物。
2.2.3 M12([M+H]+,m/z596/598)
从MDF色谱图中选择性地检测m/z596/598(m/z404/406+192 Da),在保留时间为7.25、9.02、10.46 min出现3个色谱峰,分别命名为M12-1、M12-2和M12-3,根据准确分子质量,推测它们的分子式均为C27H34ClN3O8S(原形分子式为C21H26ClN3OS),比原形多了C6H8O7。进行高碰撞能量下的全扫描质谱分析,M12-1、M12-2和M12-3产生m/z420/422、290/292、262/264、171、143、100、98碎片离子。m/z596/598→420/422为中性丢失176 Da葡萄糖醛酸分子;m/z290/292、262/264的碎片离子比原形药物的B片段多16 Da,而m/z171、143、100、98的碎片离子与原形药物的A片段相同,推测M12为M0的B部分羟基化后的葡萄糖醛酸结合物。胆汁样品经β-葡萄糖苷酸酶水解后,m/z596/598色谱峰消失,M5-2和M5-4的色谱峰明显增强,故推测M12为M5-2和M5-4的葡萄糖醛酸结合物,其提取离子流色谱图示于图3。
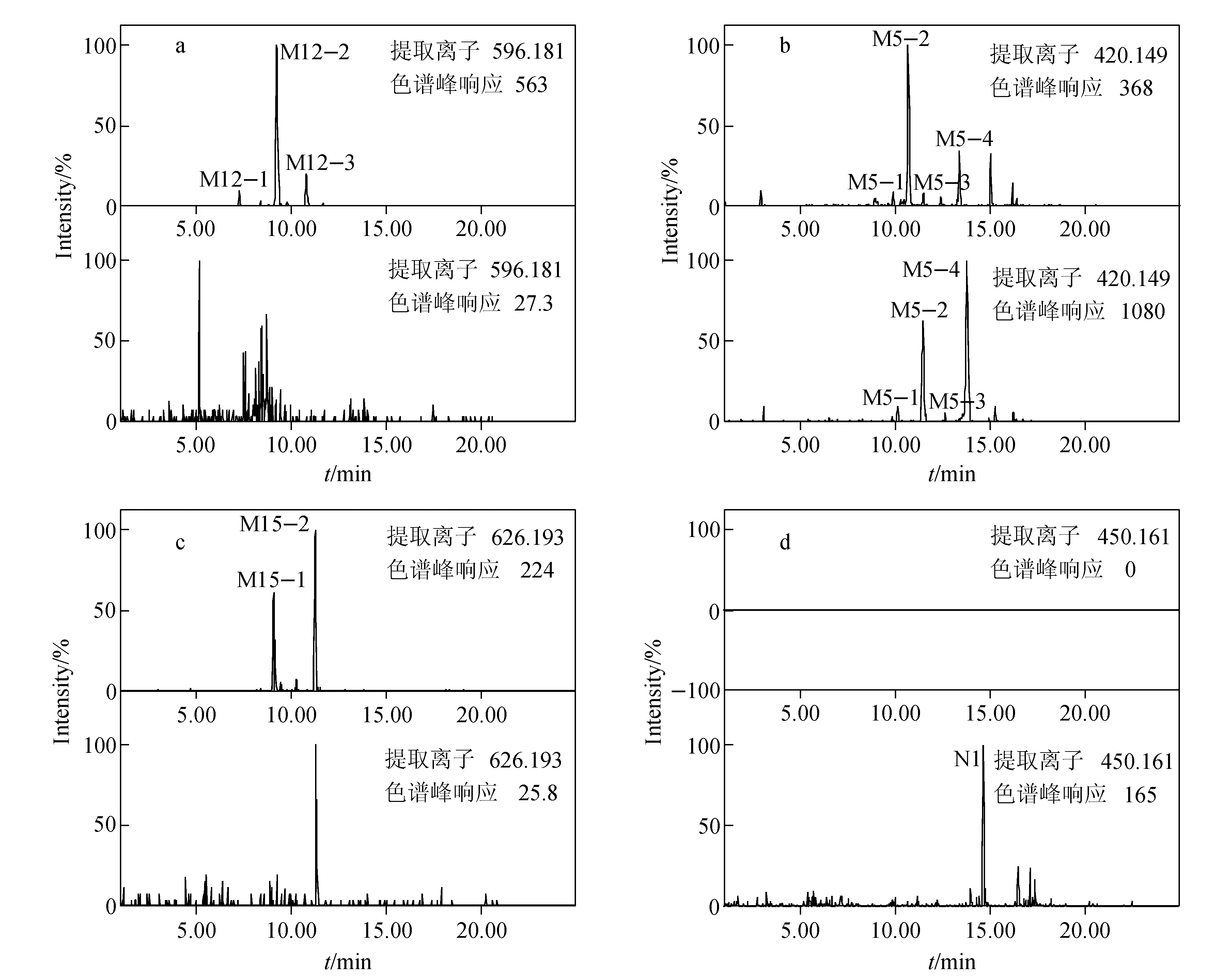
注:a.M12;b.M12的苷元M5; c.M15; d.M15的苷元N1图3 人胆汁中奋乃静代谢物经β-葡萄糖苷酸酶水解前后的提取离子流色谱图Fig.3 Extracted ion chromatograms (XICs) of perphenazine metabolites in human bile before and after treatment with β-glucuronidase
2.2.4 M15([M+H]+,m/z626/628)
从MDF色谱图中选择性地检测m/z626/628(m/z404/406+222 Da),在保留时间为8.88、10.92 min出现2个色谱峰,分别命名为M15-1和M15-2,根据准确分子质量,推测它们的分子式均为C28H36ClN3O9S(原形分子式为C21H26ClN3OS),比原形多了C7H10O8。进行高碰撞能量下的全扫描质谱分析,M15-1和M15-2产生m/z450/452、292/294、171、143、100、98的碎片离子。m/z626/628→450/452为中性丢失176 Da葡萄糖醛酸分子;m/z450/452的碎片离子比m/z404/406的原形药物多46 Da,通过准确分子质量分析,推测其分子式为C22H28ClN3O3S,比原形多CH2O2;m/z171、143、100、98的碎片离子与原形药物的A片段相同;m/z292/294的碎片离子比原形药物的B片段多46 Da,推测m/z450/452碎片离子结构可能为M0的B部分发生双羟基化后与甲基结合,M15-1和M15-2为其葡萄糖醛酸分子结合代谢物。胆汁样品经β-葡萄糖苷酸酶水解后,m/z626/628色谱峰消失,出现1个m/z450/452色谱峰,保留时间为14.10 min,命名为N1,通过准确分子质量分析,推测其分子式为C22H28ClN3O3S,且在高能量质谱图中观察到了m/z292/294、171、143、100、98的碎片离子,佐证了上述推断。
综合分析色谱及质谱数据,推测奋乃静在人体内的主要代谢途径,示于图4。代谢途径包括羟基化、脱氢、N-去烷基化、甲基化、硫酸及葡萄糖醛酸结合等。其中,原形药物和N-去烷基后双羟基化代谢物及其II相结合物、原形药物羟基化或双羟基化后脱氢及其II相结合物、原形药物双羟基化并甲基化后的II相结合物等16种代谢物均为人体内新发现的奋乃静代谢物。
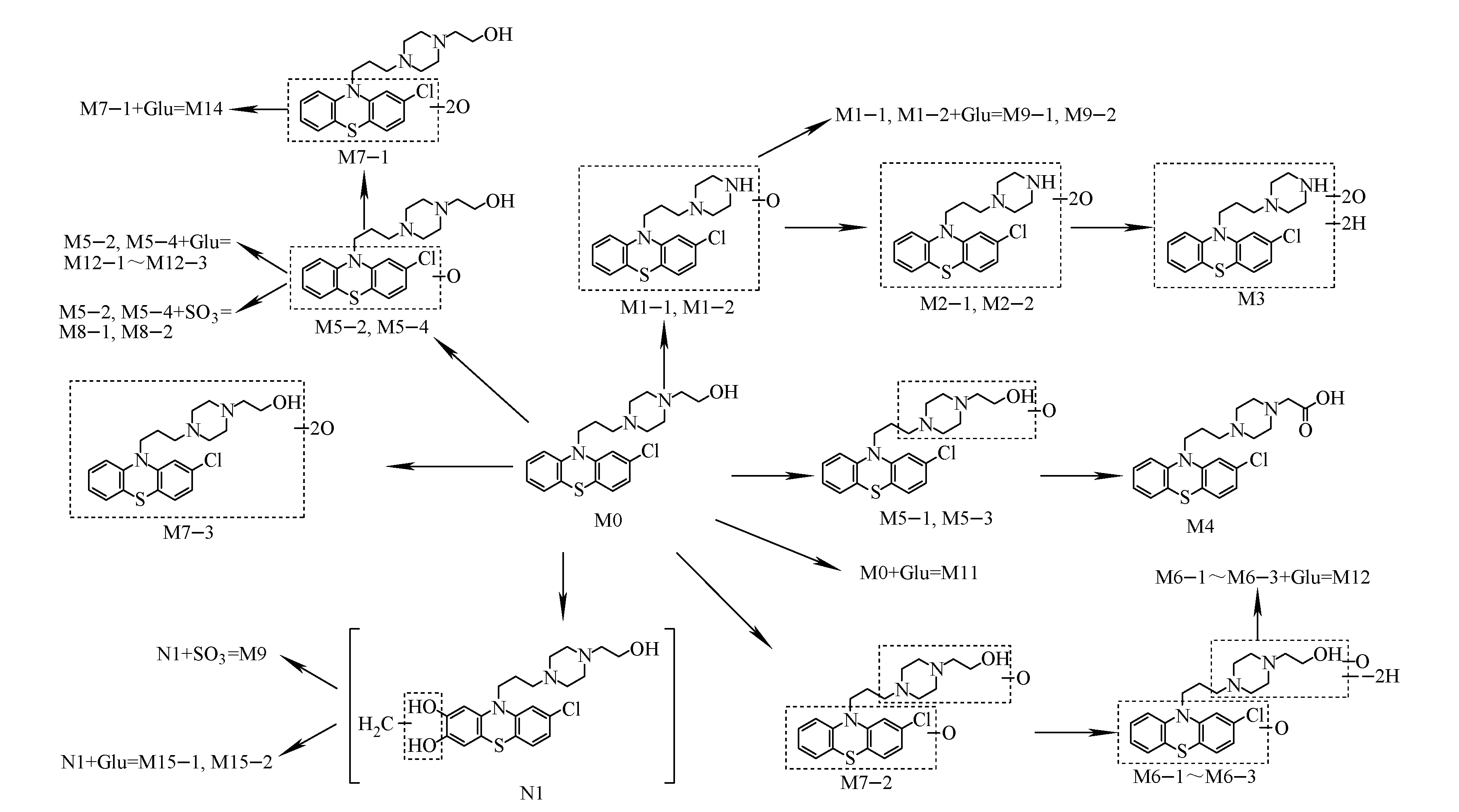
图4 推测的奋乃静在人体内的主要代谢途径Fig.4 Proposed major metabolic pathways of perphenazine in humans
3 结论
本研究采用UPLC/Q-TOF MS结合MDF和generic dealkylation等软件技术,快速检测并表征了奋乃静在人胆汁中的代谢物。患者口服奋乃静后,在人胆汁中共检测到29种代谢物,包括16种I相代谢物及13种II相代谢物,主要代谢途径为羟基化、脱氢、N-去烷基化、甲基化、硫酸及葡萄糖醛酸结合等,其中16种代谢物为首次报道的新颖代谢物。本研究鉴定了奋乃静在人胆汁中的代谢物,并进一步完善了奋乃静在人体内的代谢途径。但是,由于样品来源有限,本研究仅分析了1名受试者的胆汁样品,存在一定的局限性,后续研究可进行多样本分析,以获得更准确的实验结果。
[1] SYMCHOWICZ S, PECKHAM W D, EISLER M, et al. The distribution and excretion of radioactivity after administration of35S-labeled perphenazine (Trilafon) [J]. Biochem Pharmacol, 1962, 11(6): 417-422.
[2] EGGERT H C, ROSTED C T, ELLEY J, et al. Clinical pharmacokinetic studies of perphenazine[J]. Br J Clin Pharmacol, 1976, 3(5): 915-923.
[3] HUANG C L, KURLAND A A. Perphenazine (Tr- ilafon) metabolism in psychotic patients [J]. Arch Gen Psychiatry, 1963, 10(6): 639-646.
[4] GHIBELLINI G, LESLIE E M, BROUWER K L R. Methods to evaluate billary excretion of drugs in humans: An updated review[J]. Mol Pharm, 2006, 3(3): 198-211.
[5] ROLLINS D E, KLAASSEN C D. Biliary excretion of drugs in man[J]. Clin Pharmacokinet, 1979, 4(5): 368-379.
[6] LEVINE W G. Biliary excretion of drugs and other xenobiotics[J]. Ann Rev Pharmacol Toxicol, 1978, 18: 81-96.
[7] KLAASSEN C D, PLAA G L. Biliary excretion of xenobiotics[J]. Crit Rev Toxicol, 1975, 4(1): 1-29.
[8] DU J B, DENG P, CHEN X Y, et al. Characterization of ornidazole metabolites in human bile after intraveneous doses by ultraperformance liquid chromatography/quadrupole time-of-flight mass spectrometry[J]. Acta Pharma Sin B, 2012, 2(2): 159-167.
[9] TEICHERT J, SOHR R, HENNIG L, et al. Id- entification and quantitation of the n-acetyl-l-cysteine s-conjugates of bendamustine and its sulfoxides in human bile after administration of bendamustine hydrochloride[J]. Drug Metab Dispos, 2008, 37(2): 292-301.
[10] BURCKART G J, STARZL T E, VENKATARAMANAN R, et al. Excretion of cyclosporine and its metabolites in human bile[J]. Transplant Proc, 1986, 18(Suppl 5): 46-49.
[11] CHRISTIANS U, STROHMEYER S, KOWNATZKIJ R, et al. Investigations on the metabolic pathways of cyclosporine: I. Excretion of cyclosporine and its metabolites in human bile-isolation of 12 new cyclosporine metabolites[J]. Xenobiotica, 1991, 21(9): 1 185-1 198.
[12] WANG C P, HARTMAN N R, VENKATARAMANAN R, et al. Isolation of 10 cyclosporine metabolites from human bile[J]. Drug Metab Dispos, 1989, 17(3): 292-296.
[13] VANE F M, BUGG C J, RODRIGUEZ L C. Identification of etretinate metabolites in human bile[J]. Drug Metab Dispos, 1989, 17(3): 275-279.
[14] LUNDAHL A, LENNERNS H, KNUTSON L, et al. Identification of finasteride metabolites in human bile and urine by high-performance liquid chromatography/tandem mass spectrometry[J]. Drug Metab Dispos, 2009, 37(10): 2 008-2 017.
[15] WANG L F, ZHANG D L, SWAMINATHAN A, et al. Glucuronidation as a major metabolic clearance pathway of14C-labeled muraglitazar in humans: metabolic profiles in subjects with or without bile collection[J]. Drug Metab Dispos, 2005, 34(3): 427-439.
[16] KISANGA E R, MELLGREN G, LIEN E A. Ex- cretion of hydroxylated metabolites of tamoxifen in human bile and urine[J]. Anticancer Res, 2005, 25(6): 4 487-4 492.
[17] LIEN E A, SOLHEIM E, KVINNSLAND S, et al. Identification of 4-hydroxy-n-desmethyltamoxifen as a metabolite of tamoxifen in human bile[J]. Cancer Res, 1988, 48: 2 304-2 308.
[18] DENG P, YOU T G, CHEN X Y, et al. Identification of amiodarone metabolites in human bile by ultraperformance liquid chromatography/quadrupole time-of-flight mass spectrometry[J]. Drug Metab Dispos, 2011, 39(6): 1 058-1 069.
[19] ZHONG D F, LI X Q, WANG A M, et al. Identification of the metabolites of roxithromycin in humans[J]. Drug Metab Dispos, 2000, 28(5): 552-559.
[20] ZHU M S, MA L, ZHANG D L, et al. Detection and characterization of metabolites in biological matrices using mass defect ltering of liquid chromatography/high resolution mass spectrometry data[J]. Drug Metab Dispos, 2006, 34(10): 1 722-1 733.
[21] MORTISHIRE-SMITH R J, CASTRO-PEREZ J M, YU K, et al. Generic dealkylation: A tool for increasing the hit-rate of metabolite rationalization, and automatic customization of mass defect filters [J]. Rapid Commun Mass Spectrom, 2009, 23(7): 939-948.
[22] BATEMAN K P, CASTRO-PEREZ J M, WRONA M, et al. MSE with mass defect filtering for in vitro and in vivo metabolite identification[J]. Rapid Commun Mass Spectrom, 2007, 21(9): 1 485-1 496.
[23] MA S, CHOWDHURY S K, ALTON K B. Application of mass spectrometry for metabolite identification[J]. Curr Drug Metab, 2006, 7(5): 503-523.
猜你喜欢
食品与生物技术学报(2022年1期)2023-01-11
现代临床医学(2022年4期)2022-09-29
中草药(2022年6期)2022-03-21
中国医院用药评价与分析(2020年3期)2020-05-29
幽默大师(2019年10期)2019-10-17
阅读(快乐英语高年级)(2019年8期)2019-09-10
农药科学与管理(2019年5期)2019-08-13
分析化学(2017年12期)2017-12-25
质谱学报(2015年5期)2015-03-01
转化医学电子杂志(2015年6期)2015-01-22
