
OH自由基引发的2,3,7,8-四氯代二苯并呋喃降解机理的理论研究
2014-08-02 03:54:17孙胜敏
东北师大学报(自然科学版) 2014年4期
张 坤,孙胜敏,张 辉
(哈尔滨理工大学化学与环境工程学院,黑龙江 哈尔滨 150080)
OH自由基引发的2,3,7,8-四氯代二苯并呋喃降解机理的理论研究
张 坤,孙胜敏,张 辉
(哈尔滨理工大学化学与环境工程学院,黑龙江 哈尔滨 150080)
对2,3,7,8-四氯代二苯并呋喃(2,3,7,8-TCDF)的OH自由基加成反应机理以及后续的单分子降解反应机理进行了理论研究.从微观反应动力学角度解释了常温下2,3,7,8-TCDF作为二噁英类物质难降解的根本原因,并阐述了其在高温下的单分子降解反应机理.采用密度泛函理论,计算得到了反应势能面上各稳定点的几何结构,同时完成了简谐振动频率分析,并通过内禀反应坐标理论计算出各反应通道的最低能量路径.结果表明,OH自由基容易加成到2,3,7,8-TCDF上,但后续的降解反应的能垒过高,因此2,3,7,8-TCDF降解反应只能在高温下进行.
2,3,7,8-四氯代二苯并呋喃;二噁英;过渡态;降解机理
二噁英是一类常见的持久性有机污染物(POP),具有剧毒,能致癌、致基因突变、致畸、干扰内分泌并破坏免疫系统,其主要来源是垃圾焚烧和工业生产[1-3].在自然条件下难以降解,危害了环境安全和人类健康.目前已引起全世界科学界和公众的高度关注,因此对二噁英的生成和降解机理的研究已成为近20年来的研究热点.目前二噁英的生成机理已经被大量研究并报道,而二噁英的降解反应机理相对较复杂,未有统一明确的结论.
2008年Mohammednoor Altarawneh等人对二苯并呋喃在气相条件下的氧化和降解反应进行了理论研究[4],表明OH自由基最易加成到与氧原子毗邻的碳原子上,并且加合物能继续和O2反应,生成DF—OH—O2加合物;2012年文献[5]对DF—OH—O2加合物与大气中的NO和H2O的后续反应进行了研究,给出了2,3,7,8-TCDF的大气化学降解模型,但是只能描述二噁英类物质的大气环境中的降解,而对于焚烧过程中的二噁英类物质的降解不能很好解释.因此我们尝试对二噁英类物质在焚烧炉高温条件下的复杂热降解机理进行理论研究.实验研究表明多氯代二苯并呋喃(PCDF)比多氯代二苯并二噁英(PCDD)更稳定,更难降解,在垃圾焚烧中累积生成量相对较多[6-8],故本文研究选择2,3,7,8-TCDF作为研究对象,另外,OH自由基是燃烧焚烧过程中常见的自由基[9],而且许多研究表明OH自由基容易与二噁英类物质发生加成反应[5,10-13].因此我们主要考虑的是2,3,7,8-TCDF 的OH自由基加成反应和后续的降解反应过程.本文所研究的反应路径见图1.
2条反应通道编号为R1和R2.其中反应物标记为2,3,7,8-TCDF和OH,反应过渡态为TS1,TS1-1,TS1-1-s,TS1-2以及TS1-2-s,中间物为IM1,IM1-1和IM1-2,产物为P1-1-s和P1-2-s.

图1 反应路径
1 计算方法
量子化学计算广泛应用于对物理化学现象的微观机理研究[14-15],本文应用密度泛函理论方法[16],在B3LYP/6-311+G(d,p)计算水平下,分别优化了2,3,7,8-TCDF的OH自由基加成反应及后续的单分子反应过程中的反应物、过渡态、中间物和生成物的几何结构,并对得到的平衡几何构型进行简谐振动频率计算分析,确认所得到的几何构型为势能面上的极值点.在相同水平下,以过渡态结构为出发点,利用内禀反应坐标理论[17-18],得到了2个反应通道的最小能量路径.在MPWB1K/6-311+(3df,2p)水平下对反应势能面上所有稳定点进行高水平单点能计算,获得较准确的势能面信息.以上计算是利用Gaussian 09程序[19]在SGI 3800服务器和中科院超级计算中心的科学计算网格SCGrid上完成.
2 结果与讨论
2.1 稳定点性质
在B3LYP/6-311+G(d,p)水平下,优化了2,3,7,8-TCDF的OH自由基加成反应及后续的单分子反应中的反应物(2,3,7,8-TCDF和OH)、反应过渡态(TS1,TS1-1,TS1-1-s,TS1-2和TS1-2-s)、中间物(IM1,IM1-1和IM1-2)以及产物(P1-1-s和P1-2-s)的平衡几何构型,优化结果见图2.
反应中涉及的所有过渡态的简谐振动频率列于表1,表1中的频率均已进行校正,校正因子为0.968 8[20].数据表明过渡态TS1,TS1-1,TS1-1-s,TS1-2和TS1-2-s 均有且仅有一个虚频,都是势能面上的一阶鞍点.查看所有虚频的振动模式,均对应着相应基元反应的断键和成键.

表1 在B3LYP/6-311+G(d,p)水平下计算的过渡态的简谐振动频率 cm-1

续表1 cm-1


图2 在B3LYP/6-311+G(d,p)水平下优化得到的反应物、反应过渡态、中间物以及产物的平衡几何构型
2.2 能量


图3 在MPWB1K/6-311+(3df,2p)//B3LYP/6-311+G(d,p)双水平下所得的反应势能图
由图3可知,2,3,7,8-TCDF的OH加成反应能垒较低,加成反应容易发生.而氢迁移和异构化反应的能垒很高,可能需要在高温条件下才能发生.由于中间产物IM1-1和IM1-2的相对能量较高,反应时如果无法克服TS1-1-s和TS1-2-s的能垒而进入产物区,则很容易越过TS1-1和TS1-2而返回到IM1.
各反应步骤的反应能垒和反应焓变见表2.其中2,3,7,8-TCDF的OH基加成反应(r1)的能垒为24.27/31.59 kJ/mol,而且反应放热,反应容易进行;而后续的氢迁移反应(r2和r4)与异构化反应(r3和r5)均具有较高的反应能垒,氢迁移反应为吸热反应,而异构化反应为放热反应,但放出的热量不足以克服其相应的能垒高度.
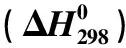
表2 各基元反应在MPWB1K/6-311+(3df,2p)//B3LYP/6-311+G(d,p)双水平下的标准摩尔反应焓和加零点振动能校正的反应能垒(ΔETS+ΔEZPE) kJ/mol
2.3 反应速率常数
为了考察不同温度下2,3,7,8-TCDF的反应情况,利用以上的势能面信息进行反应动力学计算.由于本文研究涉及的反应为多势阱反应,且考虑到实际反应条件为高温焚烧,因此采用了在多势阱反应和热化学反应计算结果比较精确的VARIFLEX代码来计算反应速率常数,计算的温度范围为298K~2 000K,计算结果绘于图4—6.其中图4为2,3,7,8-TCDF的OH加成反应速率常数随温度变化曲线;图5为IM1转化为P1-1-s的单分子反应速率常数随温度变化曲线;图6为IM1转化为P1-2-s的单分子反应速率常数随温度变化曲线.

图4 2,3,7,8-TCDF的OH加成反应速率常数随温度变化曲
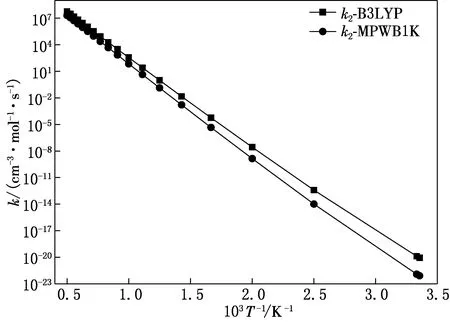
图5 IM1转化为P1-1-s的单分子反应速率常数随温度变化曲线
图4中2条曲线k1-B3LYP,k1-MPWB1K分别为2,3,7,8-TCDF的OH加成反应在B3LYP/6-311+G(d,p)和MPWB1K/6-311+(3df,2p)水平下的反应速率常数,同时文献[5]报道的298.15K时的反应速率常数也作为参考数据绘于图中.反应速率常数随温度升高而增大,且增幅逐渐减小.对比可知,B3LYP/6-311+G(d,p)水平下的反应速率常数更接近文献参考值.MPWB1K/6-311+(3df,2p)水平下得到的反应速率常数相对偏低.
由图5可知,IM1转化为P1-1-s的单分子反应的反应速率常数随温度呈正相关.且MPWB1K/6-311+(3df,2p)水平下的反应速率常数在所研究的温度范围内均低于B3LYP/6-311+G(d,p)水平下所得的反应速率常数.速率常数的差值随温度升高而减小.

图6 IM1转化为P1-2-s的单分子反应速率常数随温度变化曲线
由图6可知,反应速率常数随温度升高而增大,MPWB1K/6-311+(3df,2p)水平下的反应速率常数在所研究的温度范围内均低于B3LYP/6-311+G(d,p)水平下所得的反应速率常数.但2种水平下的反应速率常数的差值逐渐减小,在高温区二者基本相等.
通过比较2,3,7,8-TCDF的OH加成反应和加成产物IM1的单分子反应可知,在同样的温度范围内,单分子反应过程对温度更加敏感,随温度升高速率常数变化巨大,约30个数量级.而加成反应作为双分子反应其速率常数随温度升高而变化幅度较小,只有3~6数量级.即单分子反应速率常数的温度敏感性要强于2,3,7,8-TCDF的OH加成反应.从热力学角度看,OH加成反应的反应能垒低于IM1的单分子反应过程(氢迁移和异构化)的能垒,但是从动力学来看,虽然低温时加成反应速率高于单分子反应,但随着温度的升高,加成反应速率反而逐渐低于单分子反应过程速率,尤其是400K以后,单分子反应速率常数已远远超过了加成反应.
这样的热力学和动力学不一致,从微观角度来讲是由于双分子反应和单分子反应的反应物活化方式不同,双分子反应需要2个反应物分子之间的碰撞,且只有碰撞发生在能引起反应的自由度上才能算作有效碰撞,所以分子的自由度越大,有效碰撞概率越小,反应相对较慢;而单分子反应的情况是反应物与任何分子发生的碰撞都有利于反应发生,只要能发生碰撞,反应物就能获得能量被活化.在温度较低时分子动能较小,每次碰撞反应物获得的能量较少,要经过时滞过程来进行内部能量传递和分配以达到成键断键所需的能量,这就解释低温时单分子反应的反应速率较低的原因.随着温度升高,反应物在碰撞过程中能获得较多能量,一旦分子的能量足以克服反应能垒,反应就能瞬时完成,因此在高温区,单分子反应的反应速率迅猛增长,远远超过了OH加成的双分子反应速率.
为了方便估算其他温度下的反应速率常数,我们将计算得到的速率常数进行三参数拟合,得到速率常数的三参数阿伦尼乌斯表达式列于表3.

表3 B3LYP/6-311+G(d,p)和MPWB1K/6-311+(3df,2p)水平下的反应速率表达式
3 结论
本文在MPWB1K/6-311+(3df,2p)//B3LYP/6-311+G(d,p)水平下对OH自由基引发的2,3,7,8-TCDF的降解机理进行了理论计算研究.计算结果表明:
(1) 2,3,7,8-TCDF的OH加成反应能垒较低,加成反应容易发生,而加成产物IM1的氢迁移和异构化反应的能垒则相对很高,低温下反应难以进行.
(2) 通过反应动力学计算,2,3,7,8-TCDF的OH加成反应速率常数随温度升高而增大,加成产物IM1的单分子反应的速率常数也随温度升高而增大,且其温度敏感性要强于2,3,7,8-TCDF的OH加成反应.
(3) 在OH加成到2,3,7,8-TCDF的基础上,加成产物IM1能在高温下进行系列的单分子反应,从而破坏2,3,7,8-TCDF的骨架结构.
[1] KULKARNI P S,CRESPO J G,AFONSO C A M. Dioxins sources and current remediation technologies—a review[J]. Environment International,2008,34(1):139-153.
[2] BUEKENS A,STIEGLITZ L,HELL K,et al.Dioxins from thermal and metallurgical processes:recent studies for the iron and steel industry[J]. Chemosphere,2001,42:729-735.
[3] 陈彤. 城市生活垃圾焚烧过程中二噁英的形成机理及控制技术研究[D].浙江大学,2006.
[4] ALTARAWNEH M,KENNEDY E M,DLUGOGORSKI B Z,et al. Computational study of the oxidation and decomposition of dibenzofuran under atmospheric conditions[J]. The Journal of Physical Chemistry A,2008,112(30):6960- 6967.
[5] XIAOMIN SUN,CHENXI ZHANG,YUYANG ZHAO,et al. Atmospheric chemical reactions of 2,3,7,8-tetrachlorinated dibenzofuran initiated by an OH radical:mechanism and kinetics study[J]. Environmental Science & Technology,2012,46 (15):8148-8155.
[6] STIEGLITZ L,ZWICK G,BECK J,et al. On the de novo-synthesis of PCDD/PCDF on fly ash of municipal waste incinerators[J]. Chemosphere,1989,18:1219-1226.
[7] VOGG H,STIEGLITZ L. Thermal behavior of PCDD/PCDF in fly ash from municipal incinerators[J]. Chemosphere,1986,15:1373-1378.
[8] SCHWARZ G,STIEGLITZ L,ROTH W. Formation conditions of several polychlorinated compound classes on fly ash of a municipal waste incinerator[J]. Organohalogen Compounds,1990,3:169-172.
[9] WILSON JR W E.A critical review of the gas-phase reaction kinetics of the hydroxyl radical[J]. J Phys Chem Ref Data,1972,1(2):535-573.
[10] ZHANG C,ZHAO Y,BAI J,et al.Mechanism and kinetic study on the OH-initiated degradation of 2,3,7,8- tetrachlorinated dibenzofuran in atmosphere[J]. Science of the Total Environment,2012,435:53-60.
[11] ZHANG C,SUN T,SUN X. Mechanism for OH-initiated degradation of 2,3,7,8-tetrachlorinated dibenzo-p- dioxins in the presence of O2and NO/H2O[J]. Environmental science & technology,2011,45(11):4756-4762.
[12] ALTARAWNEH M,KENNEDY E M,DLUGOGORSKI B Z,et al. Computational study of the oxidation and decomposition of dibenzofuran under atmospheric conditions[J]. The Journal of Physical Chemistry A,2008,112(30):6960-6967.
[13] LEE J E,CHOI W,MHIN B J,et al.Theoretical study on the reaction of OH radicals with polychlorinated dibenzo-p-dioxins[J]. The Journal of Physical Chemistry A,2004,108(4):607-614.
[14] 张辉,李玥,赵洪,等. 苯乙酮烯醇式互换异构对聚乙烯电树枝老化性能影响的理论研究[J]. 东北师大学报:自然科学版,2012(4):97-100.
[15] 黄伣丽,李玥,张辉. OXO(X=Cl,Br)自由基与CO和NO自由基反应机理的理论研究[J]. 东北师大学报:自然科学版,2012(3):91-94.
[16] PARR R G,YANG W. Density-functional theory of atoms and molecules[M]. New York:Oxford University Press,1989:47-69.
[17] FUKUI K. The path of chemical reactions-the IRC approach[J]. Acc Chem Res,1981,14(12):363-368.
[18] 赵成大. 内禀反应坐标法及其在单分子反应中的应用[J]. 东北师大学报:自然科学版,1986(3):47-55.
[19] FRISCH M J,TRUCKS G W,CHEESEMANJ R,et al. Gaussian 09[CP]. Gaussian Inc Wallingford CT,2009.
[20] MERRICK J P,MORAN D,RADOM L. An evaluation of harmonic vibrational frequency scale factors[J]. The Journal of Physical Chemistry A,2007,111(45):11683-11700.
(责任编辑:石绍庆)
The mechanism study of the degradation reaction of 2,3,7,8-TCDF initiated by OH radical
ZHANG Kun,SUN Sheng-min,ZHANG Hui
(College of Chemical and Environmental Engineering,Harbin University of Science and Technology,Harbin 150080,China)
In this paper,the mechanism on the addition reaction of 2,3,7,8-TCDF with OH radical and subsequent unimolecular decomposition reaction are investigated by theoretical calculation. The recalcitrance of 2,3,7,8-TCDF at room temperature is explained from the perspective of microscopic kinetic,and the mechanism of unimolecular degradation reaction at high temperature is expounded simultaneously. In this study,the density functional theory is employed to calculate the equilibrium geometries of the stable points on the reaction potential energy surface,and the harmonic vibration frequencies analysis is completed. The minimum energy path (MEP) is obtained by The intrinsic reaction coordinate (IRC) theory. The study results show that the OH radical addition reaction of 2,3,7,8-TCDF can occur easily,but the degradation reaction of 2,3,7,8-TCDF can only happen at high temperatures due to the high energy barrier of subsequent unimolecular degradation reaction.
2,3,7,8-TCDF;dioxin;transition state;degradation mechanism
1000-1832(2014)04-0103-07
10.11672/dbsdzk2014-04-020
2014-07-10
教育部博士点专项基金资助项目(20112303110005);黑龙江杰出青年基金资助项目(JC201206).
张坤(1989—),男,硕士研究生;通讯作者:张辉(1967—),女,教授,博士研究生导师,主要从事分子反应动力学机理研究.
O 641.1 [学科代码] 150·30
A
猜你喜欢
燃料化学学报(2023年3期)2023-03-11 03:34:40
北京航空航天大学学报(2022年5期)2022-06-06 09:27:18
大学化学(2021年8期)2021-09-26 10:51:16
中学课程辅导·教学研究(2021年8期)2021-07-14 13:44:52
数学年刊A辑(中文版)(2021年1期)2021-06-09 09:32:06
燃料化学学报(2021年5期)2021-06-02 14:01:38
电脑知识与技术(2018年3期)2018-03-21 09:27:04
哈尔滨理工大学学报(2017年1期)2017-04-08 04:16:24
山西大同大学学报(自然科学版)(2016年4期)2016-11-27 02:20:55
新高考·高一物理(2016年3期)2016-05-18 16:16:56
