
基于LibSVM的CKSAAP蛋白特征提取预测水稻蛋白质磷酸化位点*
2014-07-14 05:49何华勤
湖北科技学院学报 2014年7期
王 伟,何华勤
(福建农林大学,福建 福州 350002)
引言
由于蛋白质领域研究的日益进步以及基因测序、编码技术的普及,各大数据库中已经大量收集了各种蛋白质的氨基酸序列。因为蛋白质组学研究的重要领域是蛋白质功能,因此研究蛋白质序列已经成为生物信息学中不可或缺的部分[1][3]。Vapnik和Cortes于1995年首先提出支持向量机(全名Support Vector Machine)这一概念,它的基本原理是在线性可分的基础上,通过自身的算法将线性可分变为线性不可分[2]。通过此转变我们可以在非线性函数中进行使用和计算,这种分类算法被称为支持向量机,即SVM。将支持向量机算法应用到水稻蛋白质磷酸化位点的预测当中去,是现在研究水稻蛋白质磷酸化的一个重要方向。
一、基于氨基酸组成的特征提取算法
将一条蛋白质表示为S=R1R1R2R3…RL,其中Ri表示蛋白质序列中 i个位置上的氨基酸,L表示的是蛋白质序列的长度;20种氨基酸用单字母表示如下:
AA={A,C,D,E,F,G,H,I,K,L,M,N,P,Q,R,S,T,V,W,Y}
CKSAAP的定义如下:
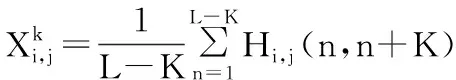
(1)
其中 i和j 各自表示二十种各不相同的氨基酸;若序列中 n位置上为氨基酸,并且位置n+K 同为氨基酸j时, Hi,j(n,n+K)=1;否则为零。序列片断中残基对的K个间隔的组成情况通过该编码来表示,同时展示了序列或者序列片断间残基的小范围互作[4]。
而序列片段中K个间隔的残基对个数我们用Vi 来表示,特征向量的维数用i来表示。对应K的取值各不相同,那么i也会相应变化。在特征值的转换过程中,把每条序列的Vi值分别算出,即在序列中每个残基对的总数[5]。
特征向量被定义为:
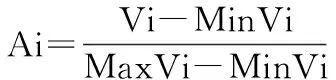
(2)
其中,Ai表示特征值, MaxVi为序列中Vi的最大值, MinVi为序列中Vi的最小值。
二、模型的建立与评估
根据上述CKSAAP算法原理,我们利用Python编程环境,将从swiss-prot下载下来进行整理的序列转换成Libsvm可以识别的格式。构建完数据集合之后,我们选取部分数据来进行训练。对于要进行预测的数据,我们分别从总数据集合的数据中随机抽取十次正负样本,选取的正负样本比例为1:1。
在利用libSVM进行预测之前,使用交叉验证对所提取的特征值进行评估和测试,得到不同的Cost值和Gamma值后,从中选取模型所需的最优参数。通过比对我们选取rbf核类型和c-svc类型来创建模型。SVM中模型是通过正负样本集来构建的,并且正负样本比例为1:1。对于易为磷酸化的S(丝氨酸)、T(苏氨酸)和Y(酪氨酸)的子集,分别从相应总训练集的正负位点数据中随机抽取十次正负样本[6]。
分别对每个序列子集的10个SVM模型进行交叉验证,通过对结果的比对和分析分别从中选取交叉验证性能最高的模型作为SVM的子模型。通过libsvm中的grid.py进行参数优选得出最优参数训练出最终模型。再通过此模型,应用svm_predict进行预测。
预测结果:
参数优选中的最佳准确率accuracy=80.2218%,而实际中预测的准确率为80.638%。
三、不同预测方法的性能比较
磷酸化位点预测工具有很多,本文应用自己构建的测试数据集来对本文工具与PlantPhos和Musite的预测性能进行对比。
我们将本文的预测工具和PlantPhos、Musite对同一测试集数据进行预测,首先将数据分成1:1的正负样本集,即磷酸化和非磷酸化位点。然后算出这三种方法的Sn(灵敏度)、Sp(特异性)、ACC(准确度)和MCC(马修斯系数)来比较各自的预测性能。
由表1可知,本文的预测工具对丝氨酸预测的准确性ACC和马修斯系数MCC分别为80.8%和0.621,plantPhos的ACC为61.2%和MCC为0.311,而Musite预测的ACC和MCC分别为72.1%和0.426。而本文的预测工具对苏氨酸位点预测的准确性ACC和马修斯系数MCC分别为79.9%和0.597,plantPhos的ACC为59.3%和MCC为0.276,而Musite预测的ACC和MCC分别为60.2%和0.206。表明本文的预测工具对磷酸化丝氨酸、苏氨酸位点的预测性能高于PlantPhos及Musite。
本文的预测方法在预测酪氨酸位点的ACC和MCC分别为81.3%和0.616,显著高于PlantPhos的57.0%和0.182,以及Musite的ACC值50%。说明本文的预测工具对磷酸化酪氨酸位点的预测性能显著高于PlantPhos及Musite。
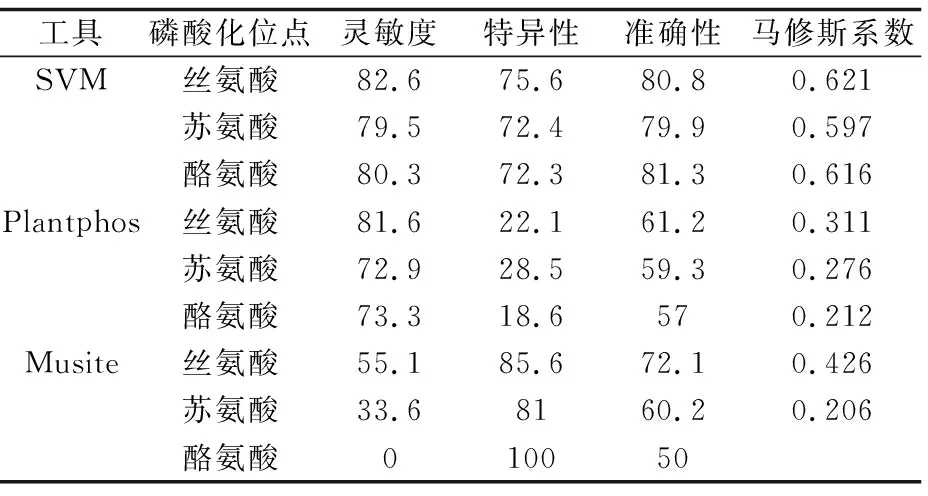
表1 预测结果对比表
参考文献:
[1] 赵凌志,刘颖,等.Weighted SVM在蛋白质磷酸化位点预测中的应用[D].北京:清华大学软件学院,2006.
[2] 蔡津津.蛋白质磷酸化位点预测与规则抽取方法研究[D].北京:中国科学院计算技术研究所, 2006.
[3] 张颖,罗辽复,吕军.使用多样性增量预测磷酸化位点[J].内蒙古大学学报(自然科学报),2008,(1).
[4] 朱玉贤,李毅,郑晓峰.现代分子生物学(第三版)[M].北京:高等教育出版社,2011.
[5] 白海燕,吕军,张颖,等.蛋白质磷酸化位点的识别[J].内蒙古工业大学学报,2011,(2).
[6] Koenig M ,Grade N. Highly specific prediction of phosphorylation sites in proteins[J], Bioinformatics, 2004.
猜你喜欢
生物化学与生物物理进展(2022年6期)2022-07-21
中学生数理化(高中版.高考理化)(2021年2期)2021-03-19
天津医科大学学报(2019年6期)2019-08-13
中国洗涤用品工业(2019年4期)2019-05-11
中成药(2018年1期)2018-02-02
分析化学(2017年12期)2017-12-25
中成药(2017年3期)2017-05-17
池州学院学报(2015年3期)2016-01-05
安徽医科大学学报(2015年9期)2015-12-16
动物医学进展(2015年10期)2015-12-07
