
斯蒂芬酸/1,4-二氧六环溶剂化物晶体结构的表征与计算
2014-06-27 05:41:50陈鹏源张琳朱顺官程广斌
兵工学报 2014年10期
陈鹏源,张琳,朱顺官,程广斌
(南京理工大学化工学院,江苏南京 210094)
斯蒂芬酸/1,4-二氧六环溶剂化物晶体结构的表征与计算
陈鹏源,张琳,朱顺官,程广斌
(南京理工大学化工学院,江苏南京 210094)
为探索共晶炸药形成机理,采用蒸发溶剂法制备了斯蒂芬酸/1,4-二氧六环溶剂化物。用单晶X射线衍射对其结构进行了表征。结构分析表明1,4-二氧六环上的氧和相邻的两个斯蒂芬酸分子中的羟基各自形成—OH…O强氢键,1,4-二氧六环上的氢和斯蒂芬酸上硝基中的氧形成CH…O—NO弱相互作用,这些作用是形成该溶剂化物的主要驱动力。分别计算了斯蒂芬酸和斯蒂芬酸/1,4-二氧六环溶剂化物晶体的Mulliken电荷和态密度。计算结果表明:形成溶剂化物后,斯蒂芬酸上的硝基电荷变多;p轨道对溶剂化物的形成起到重要的作用;斯蒂芬酸中的氢和1,4-二氧六环中的氧存在杂化。
兵器科学与技术;溶剂化物;晶体;分子间作用力;氢键
0 引言
发展高能钝感含能材料一直是人们追求的目标,目前研究的方向主要是新的单质炸药的合成和对现有炸药的改性[1]。改变炸药晶体形貌或包覆等改性取得的效果十分有限。炸药共晶可以从分子层面改变晶体结构,从而改变炸药的熔点、感度和爆炸性能等,因此这种设计思想受到越来越多的关注[2]。
Landenberger等制备了TNT和17种芳香族化合物的系列共晶,认为π-π作用是炸药共晶的组装方式[3]。Bolton等[4]获得了CL-20/TNT共晶炸药,发现形成共晶的主要作用力是CL-20硝基上的氧与TNT的氢间的氢键,TNT中缺电子的苯环与CL-20中硝基之间的静电作用对共晶形成也有贡献。该共晶大大降低了CL-20感度。Millar等[5]报道了CL-20和4种溶剂的共晶,通过结构分析认为,CL-20的4种共晶中缺少氢键的作用,而CL-20分子结构的适应性是形成共晶的重要因素,且共晶中CL-20分子的构型和去溶剂化后的构型之间存在联系。Bolton等[6]还获得了HMX/CL-20共晶,其计算爆速大于HMX,实测感度与HMX相当。Bolton等在最新研究中又提出了富电子的π堆积和缺电子的芳香环之间作用是构成共晶的一种新方式。Lin等制备了HMX和3种溶剂的共晶,结构分析和理论计算表明HMX上的亚甲基有很高的化学活性,容易和吸电子基团形成氢键[7-8]。上述研究表明炸药共晶是一种改善炸药感度及爆炸性能的手段,但对其形成机理的认识还有待进一步深入和发展,这就需要开展多种共晶炸药的制备和理论研究,以形成有效的共晶炸药设计方法。
三硝基间苯二酚又称斯蒂芬酸,是一种常见的硝基酚类炸药[9]。它的铅盐,即斯蒂芬酸铅用作起爆药。实验发现斯蒂芬酸可以和1,4-二氧六环形成溶剂化物。根据以往的研究,硝基苯类炸药较为容易形成共晶,因此本文通过对斯蒂芬酸/1,4-二氧六环溶剂化物的实验和理论研究,进一步掌握此类共晶的形成规律,为共晶炸药的设计和制备提供指导。
1 实验与理论计算方法
1.1 溶剂化物的制备
1 g斯蒂芬酸溶解于25 mL 1,4-二氧六环中,搅拌30 min后过滤。母液放入25℃恒温培养箱中蒸发结晶,3 d后得到可以用于结构测定的无色柱状晶体。
1.2 晶体结构测定
选取0.25 mm×0.22 mm×0.17 mm的单晶,在德国布鲁克公司的Bruke SMART-APEX CDD单晶衍射仪上测试。Mo-Ka射线(波长λ=0.710 73 Å),石墨单色器,3.70°≤θ≤59.98°范围,ω扫描收集得到数据。
1.3 理论计算方法
为了研究斯蒂芬酸/1,4-二氧六环溶剂化物的形成机理以及溶剂分子对斯蒂芬酸的影响,对斯蒂芬酸及其溶剂化物都进行计算。为了确保计算结果的可信度,两种晶体的计算采用同样的方法及精度。所有的计算都在Materials Studio软件CASTEP[10]模块中完成。
运用密度泛函(DFT)方法和平面波基组对上述两种晶体进行周期性计算。离子和电子的相互作用采用超软Vanderbilt恒赝势方法加以描述,晶体结构的弛豫采用BFGS方法。采用GGA/PBE泛函。平面波基组的截断能取600 eV.用于斯蒂芬酸、斯蒂芬酸/1,4-二氧六环溶剂化物计算的K点网格分别取1×1×1和1×2×1.
在构型优化时,能量收敛精度为2.0×10-5eV/Å;作用在每个原子上最大力收敛精度为0.05 eV/Å;最大位移收敛精度为0.002 Å;最大应力收敛精度为0.1 GPa.
2 结果与讨论
2.1 晶体结构
晶体结构用SHELXL-97[11]程序中基于F2的全矩阵最小二乘法进行精修。所得晶体数据已收录于剑桥晶体学数据库(CCDC号为965 983).晶体结构数据见表1.晶体的晶胞堆积图和不对称结构单元图见图1和图2.
从不对称结构单元图中可以看出,斯蒂芬酸/1, 4-二氧六环溶剂化物是由一分子斯蒂芬酸和一分子1,4-二氧六环组成。从晶胞堆积图中可以看出晶体中存在大量的氢键。
从图3中可以看出:1,4-二氧六环上的氧和相邻的两个斯蒂芬酸分子中的羟基各自形成—OH…O强氢键,氢键键长分别为1.986 Å和2.080 Å.1, 4-二氧六环上的氢和斯蒂芬酸上硝基中的氧形成CH…O—NO弱相互作用,键长为2.817 Å和2.684 Å.斯蒂芬酸分子内也形成—OH…O—NO氢键,键长分别为1.996 Å和2.157 Å.从b轴看,1,4-二氧六环和两边的斯蒂芬酸分子通过氢键连接起来,无限延伸,这点从晶胞堆积图中也可以看出。氢键是形成斯蒂芬酸/1,4-二氧六环溶剂化物的主要驱动力。晶体中分子间和分子内氢键作用力的长度和角度见表2.

表1 斯蒂芬酸/1,4-二氧六环溶剂化物晶体结构数据Tab.1 Crystal data of styphnic/1,4-dioxane solvate
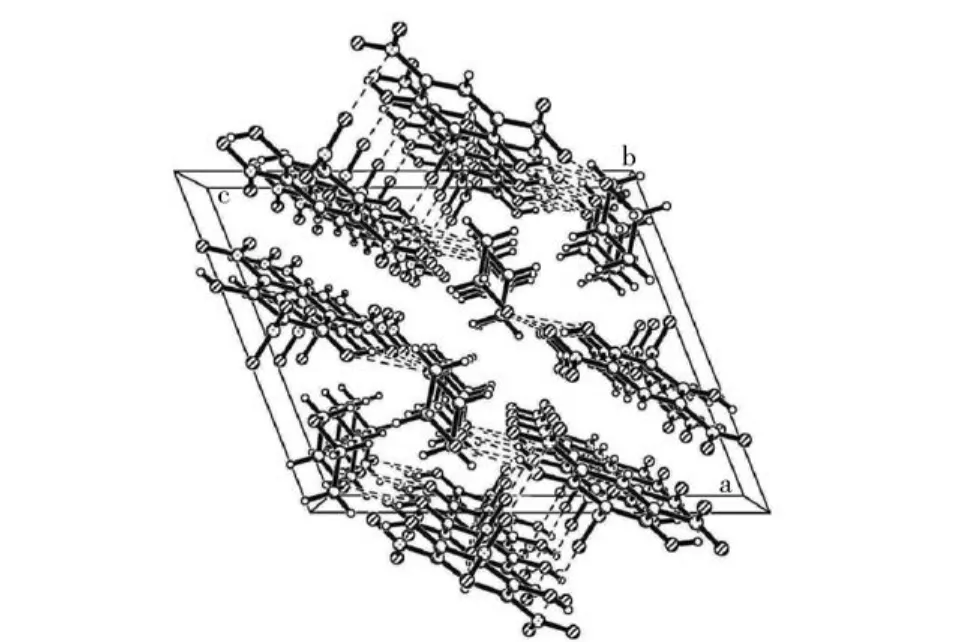
图1 晶胞堆积图Fig.1 Crystal packing of solvate
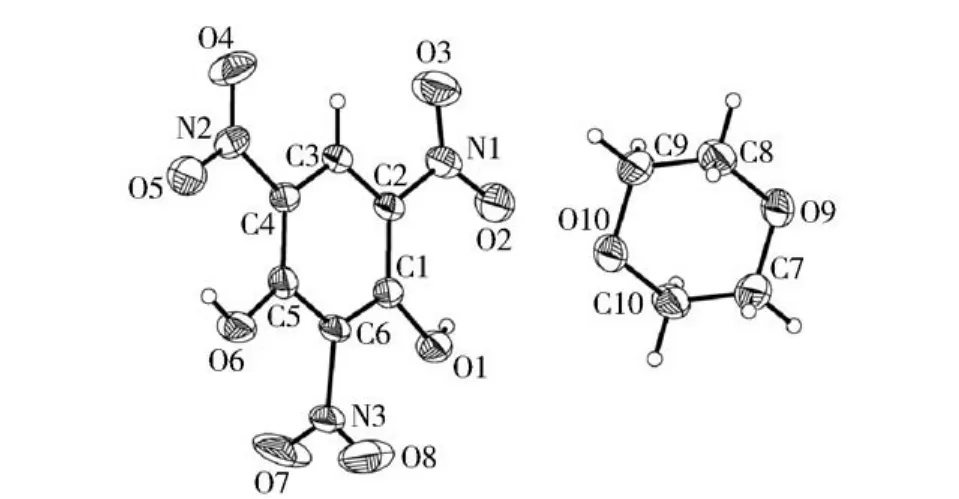
图2 不对称结构单元图Fig.2 Molecule structure of solvate

图3 斯蒂芬酸/1,4-二氧六环溶剂化物分子间和分子内氢键作用力Fig.3 The intermolecular and intramolecular hydrogen bond interaction of styphnate/1,4-dioxane solvate

表2 斯蒂芬酸/1,4-二氧六环溶剂化物晶体中的分子间作用力的的长度和角度Tab.2 Intermolecular interaction distances and angles of styphnate/1,4-dioxane solvate
Hirshfeld面和指纹图可以定性和定量的分析晶体中分子间作用力[12]。Hirshfeld面由晶体中电子分布构成,它到外部或内部最近原子的距离分别用de和di表示。标准化接触距离指范德华力。二维指纹图由de和di构成。
图4为晶体中斯蒂芬酸分子的Hirshfeld面。从图中可以看出:1,4-二氧六环中的O和斯蒂芬酸中的OH对应的Hirshfeld面颜色最深,表明作用距离最短、作用力最强,它们形成了—OH…O强氢键。斯蒂芬酸中的羟基和相邻的斯蒂芬酸分子中的硝基形成了CH…O弱氢键。这些分子间作用力分析与图3以及表2中PLATON软件分析的结果一致。Hirshfeld面对于一些较弱的作用力如O…O、C…H等也能很好地体现出来。而单纯从晶体结构分析,这些作用力不易被察觉。
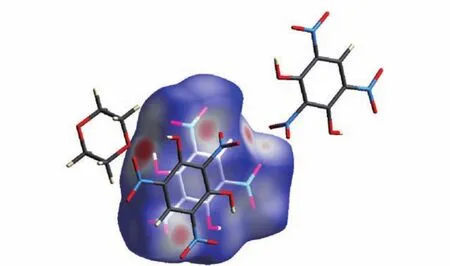
图4 溶剂化物中斯蒂芬酸分子的Hirshfeld面Fig.4 Hirshfeld surface of styphnate molecular in solvate Hirshfeld

图5 斯蒂芬酸/1,4-二氧六环溶剂化物指纹图Fig.5 The fingerprint plots of styphnate/1,4-dioxane solvate
晶体结构的分析可以定性地分析晶体中作用力的构成,而指纹图则可以非常直观地观察到分子间作用力的类型和组成。从图5和图6中可以看出, O…H和H…O作用所占比例最大,为52.2%.它对应指纹图中左下角两个分裂的尖峰,de和di值最小,即作用距离最短、作用力最强。说明氢键是形成该溶剂化物的主要驱动力,这与上文中晶体结构的分析结果一致。O…O作用占总作用力的18.0%,主要表现为相邻斯蒂芬酸中硝基之间的作用。其他一些作用主要有O…C和C…O、C…H和H…C.

图6 不同相互作用指纹图Fig.6 The fingerprint plots of different interactions
2.2 几何优化结果
晶体优化结果见表3.斯蒂芬酸a、b、c误差分别为0.82%、0.82%、-0.18%.斯蒂芬酸/1,4-二氧六环溶剂化物a、b、c、β误差分别为-1.77%、0.94%、0.26%、0.09%.二者计算误差都很小,表明计算选择的方法及参数是合理的。

表3 斯蒂芬酸与斯蒂芬酸/1,4-二氧六环溶剂化物晶体优化后结果和实验值Tab.3 Calculated and experimental lattice parameters for styphnate、styphnate/1,4-dioxane solvate
2.3 电荷分布
为了得到形成溶剂化物时电子的转移情况,分别计算了斯蒂芬酸和斯蒂芬酸/1,4-二氧六环溶剂化物的Mulliken电荷分布,计算结果见表4.

表4 斯蒂芬酸、斯蒂芬酸/1,4-二氧六环溶剂化物晶体中的Mulliken电荷分布Tab.4 Mulliken charge distribution of styphnate and styphnate/1,4-dioxane solvate
从表4可以看出,斯蒂芬酸分子在形成溶剂化物前后电荷变化的主要是O6、O7、C5、C6.O6、O7电荷从-0.57 e增加到-0.60 e,而C5、C6则分别由0.09 e减少到0.07 e、0.33 e减少到0.30 e.这表明由于1,4-二氧六环上的O对斯蒂芬酸上羟基的静电吸引作用使得斯蒂芬酸部分原子上的电荷发生转移。由此使得硝基电荷发生改变,C1、C3、C5上硝基总的硝基电荷从-0.33 e、-0.33 e和-0.17 e分别变为-0.34 e、-0.36 e和-0.23 e.根据以往的研究,硝基上的负电荷越多,分子越稳定[14]。据此可以推测,溶剂化物的感度低于斯蒂芬酸。
2.4 态密度
斯蒂芬酸/1,4-二氧六环溶剂化物C、N、O、H态密度(DOS)和总体态密度(TDOS)图见图7.在晶体的态密度图中,费米能级用虚线表示。DOS在费米能级处有一定数值,这是由于DOS中包含了一些加宽效应[15]。从图7可以看出,斯蒂芬酸/1,4-二氧六环溶剂化物晶体电子结构的一些特点:1)价带顶部主要由C、O和部分H构成,且这些峰主要来自p态的贡献;2)导带的底部主要由C、N和O的p态重叠而成,说明p轨道在化学反应中起很重要的作用; 3)TDOS价带顶部的峰连在一起,表明体系中电子的离域性很好。

图7 斯蒂芬酸/1,4-二氧六环溶剂化物局部DOS和TDOSFig.7 PDOS and TDOS of styphnate/1,4-dioxane solvate
为了分析斯蒂芬酸和1,4-二氧六环在晶体中的相互作用,计算了部分原子的DOS图。从图8中可以发现以下一些特征:在价带顶部-5~0 eV,1, 4-二氧六环中的O2p和斯蒂芬酸羟基上的H1s存在交叠,预示着它们之间存在相互作用;同样地,1, 4-二氧六环中的H1s和斯蒂芬酸硝基上的O2p存在交叠,但交叠程度相对较小,说明二者依然存在相互作用,但相对1,4-二氧六环中的O2p和斯蒂芬酸羟基上的H1s作用,其作用有所减弱。这与晶体结构的分析是一致的,1,4-二氧六环上的氧和相邻的两个斯蒂芬酸分子中的羟基各自形成—OH…O强氢键,而1,4-二氧六环上的氢和斯蒂芬酸上硝基中的氧形成CH…O—NO弱相互作用,前者的作用大于后者。此外,斯蒂芬酸分子中硝基中的O和羟基中的H在价带顶部靠近费米能级处也存在交叠,预示着分子内氢键的存在。

图8 晶体中部分原子的态密度图Fig.8 The DOS of some particular atoms in crystal
3 结论
本文制备了斯蒂芬酸/1,4-二氧六环溶剂化物并对其结构进行了表征,结构分析表明氢键是形成溶剂化物的主要驱动力。在实验结果的基础上还对其进行了理论计算。计算结果表明:形成溶剂化物后由于电荷的转移,斯蒂芬酸中硝基电荷变多,因此变得钝感。由态密度分析可知p轨道在溶剂化物的形成中起到很重要的作用,态密度分析也同时印证了氢键对形成溶剂化物的作用。
(References)
[1] 林鹤,张琳,朱顺官,等.HMX/FOX-7共晶炸药分子动力学[J].兵工学报,2012,33(9):1025-1030.
LIN He,ZHANG Lin,ZHU Shu-guan,et al.Molecular dynamic simulation of cyclotetramethylene tetranitramine/1,1-diamino-2, 2-dinitroethylene cocrystal explosives[J].Acta Armamentarii, 2012,33(9):1025-1030.(in Chinese)
[2] 郭长艳,张浩斌,王晓川,等.共晶炸药研究进展[J].材料导报,2012,26(10):49-53.
GUO Chang-yan,ZHANG Hao-bing,WANG Xiao-chuan,et al. Research progress of cocrystal explosive[J].Materials Review, 2012,26(10):49-53.(in Chinese)
[3] Landenberger K B,Matzger A J.Cocrystal engineering of a prototype energetic material:supramolecular chemistry of 2,4,6-trinitrotoluene [J].Crystal Growth&Design,2012,10(12):5341-5347.
[4] Bolton O,Matzger A J.Improved stability and smart-material functionality realized in an energetic cocrystal[J].Angewandte Chemie Intermational Edition,2011,50(38):8960-8963.
[5] Millar D I A,Maynard-Caely H E,Allam D R,et al.Crystal engineering of energetic materials:cocrystals of CL-20[J].Cryst Eng Comm,2012,14(10):3742-3749.
[6] Bolton O,Matzger A J,Pagoria P E,et al.High power explosive with good sensitivity:A 2:1 cocrystal of CL-20:HMX[J].Crystal Growth&Design,2012,12(9):4311-4314.
[7] Lin H,Zhang L,Zhu S G,et al.Synthesis,characterization,AIM and NBO analysis of HMX/DMI cocrystal explosive[J].Journal of Molecular Structure,2013,1048:339-348.
[8] Lin H,Zhang L,Zhu S G,et al.Synthesis and first principles investigation of HMX/NMP cocrystal explosive[J].Journal of Energetic Materials,2013,31(4):261-272.
[9] Kohler J,Meyer R.Explosive[M].New York:VCH,2007.
[10] Segall M,Probert M,Pickard C,et al.First principles methods using CASTEP[J].Zeitschrift fuer Kristallographie,2005,220: 567-570.
[11] Sheldrick G M.Foundations of crystallography[J].Acta Crystallographica Section A,2008,64:112-122.
[12] Spackman M A,Jayatilaka D.Hirshfeld surface analysis[J]. Cryst Eng Comm,2009,11:19-32.
[13] Pierce-Butler M A.Structures of 2,4,6-trinitro-1,3-benzenediol 2/3-hydrate and 2,4,6-trinitro-1,3,5-benzenediol 2/3-hydrate[J]. Acta Crystallogr:Sect B,1982,38:3097-3100.
[14] Zhang C Y.Review of the establishment of nitro group charge method and its applications[J].J Hazard Mater,2009,161(1): 21-28.
[15] Zhu W H,Zhang X W,Wei T,et al.First-principles study of crystalline mono-amino-2,4,6-trinitrobenzene,1,3-diamino-2,4, 6-trinitrobenzene,and 1,3,5-triamino-2,4,6-trinitrobenzene[J]. Journal of Molecular Structure:THEOCHEM,2009,900(1): 84-89.
The Crystal Structure and Theoretical Study of Styphnic/1,4-dioxane Solvate
CHEN Peng-yuan,ZHANG Lin,ZHU Shun-guan,CHENG Guang-bin
(School of Chemical Engineering,Nanjing University of Science and Technology,Nanjing 210094,Jiangsu,China)
In order to explore the mechanism of co-crystal explosive formation,styphnic/1,4-dioxane solvate was prepared by solvent evaporation method.This solvate structure was characterized by single crystal X-ray diffraction.Structural analysis shows that O in 1,4-dioxane forms—OH…O strong hydrogen bond with OH in styphnic.Moreover,H in 1,4-dioxane forms CH…O—NO weak intermolecular interaction with nitro group in styphnic.These two kinds of interactions are the main driving force for solvate composing.The Mulliken charge and density of state of both styphnic and styphnic/1,4-dioxane solvate are calculated.The result shows that the nitro group has more negative charge in the solvate than that in the pure styphnate.P orbital plays an important role in the buildup of the solvate and H in styphnate hybridizes with O in 1,4-dioxane.
ordnance science and technology;solvate;crystal;intermolecular interaction;hydrogen bond
TG156
A
1000-1093(2014)10-1569-06
10.3969/j.issn.1000-1093.2014.10.008
2013-12-13
国家自然科学基金项目(61106078)
陈鹏源(1988—),男,博士研究生。E-mail:chenpengyuan@outlook.com;张琳(1976—),女,副研究员,硕士生导师。E-mail:zhangl@mail.njust.edu.cn
猜你喜欢
山东化工(2020年9期)2020-06-01 06:56:40
疯狂英语·初中天地(2019年5期)2019-10-18 00:58:00
乐府新声(2019年3期)2019-10-17 02:17:34
化工管理(2017年32期)2017-11-24 06:20:21
NBA特刊(2017年10期)2017-08-08 03:01:46
疯狂英语·初中天地(2017年5期)2017-06-05 09:26:50
绿色科技(2017年8期)2017-05-22 21:06:55
中学生数理化·高三版(2016年2期)2016-09-10 07:22:44
中国卫生(2014年5期)2014-11-10 02:11:32
机械与电子(2014年2期)2014-02-28 02:07:43
