
食管癌细胞Fca9706 DNA聚合酶β基因敲除载体的构建
2014-06-23 16:23郭文涛路武豪孙萨迦董子明
肿瘤基础与临床 2014年3期
冯 龙,郭文涛,路武豪,孙萨迦,董子明
(1.河南中医学院基础医学院,河南郑州450008;2.郑州大学基础医学院,河南郑州450001;3.漯河医学高等专科学校,河南漯河462002;4.郑州大学第一附属医院,河南郑州450052)
食管癌细胞Fca9706 DNA聚合酶β基因敲除载体的构建
冯 龙1,2,郭文涛3,路武豪4,孙萨迦2,董子明2
(1.河南中医学院基础医学院,河南郑州450008;2.郑州大学基础医学院,河南郑州450001;3.漯河医学高等专科学校,河南漯河462002;4.郑州大学第一附属医院,河南郑州450052)
目的构建食管癌细胞Fca9706 DNA聚合酶β(DNA polβ)基因敲除载体,为DNA polβ基因敲除奠定基础。方法应用体细胞基因敲除技术的方法和原理,根据DNA polβ基因序列,设计并合成2对特异性引物(UP1/UP2和DOWN1/DOWN2),通过PCR扩增获得上游同源序列(UP)和下游同源序列(DOWN),其中上游同源序列长1 268 bp,下游同源序列长2 150 bp,将其插入骨架载体pcDNA3.1,从而构建了DNA polβ基因敲除载体pOUT-polβ,最后用PCR、酶切和测序进行鉴定。结果经过PCR筛选,限制性酶切及DNA测序鉴定,证实上游同源序列(UP)和下游同源序列(DOWN)2个片段插入正确。结论通过本研究所述方法,成功构建了用于食管癌细胞Fca9706 DNA polβ基因敲除载体pOUT-polβ。
基因敲除;DNA聚合酶;打靶载体;Fca9706细胞
DNA聚合酶β(DNA polymerase β,DNA polβ)是真核细胞DNA聚合酶家族的一员,为一看家基因,基因全长35 000 bp,位于第8号染色体近着丝点处,是单拷贝基因,为断裂基因,有14个外显子及13个内含子,转录1 400 bp mRNA,其启动子位于主要转录起始点的上游115 bp内[1]。从酵母到哺乳类细胞的polβ均高度保守,并且广泛分布于哺乳动物细胞核内,其表达产物是相对分子质量为39 000的单链小分子蛋白,是真核细胞中相对分子质量最小的DNA聚合酶。其主要功能是参与DNA损伤修复,尤其是碱基切除修复(base-excision repair,BFR),包括短片段BFR和长片段BFR[2]。
自从重组全DNA技术克隆出了DNA polβ的cDNA以后,其生物学功能越来越受到人们的关注。近年来许多学者对DNA polβ在肿瘤组织及细胞系中的突变及表达状态等进行了系统研究,为揭示肿瘤发生的分子机制,提供了一个新的切入点。目前已经在结肠癌、食管癌、乳腺癌[3]、胃癌[4]、膀胱癌[5]、前列腺癌[6-7]等多种人类肿瘤组织中发现了DNA polβ的基因突变和(或)表达异常[8-13]。
基因敲除是在20世纪80年代发展起来的一项重要的分子生物学实验技术,其利用了基因转移和重组的方法,将外源DNA导入打靶细胞中,然后将外源DNA的序列与打靶细胞染色体上同源的DNA序列进行重组,从而能够精细定点修饰以及改造目的基因片段。近年来随着分子生物学技术的不断发展,基因打靶也在不断完善,并且,在干细胞基因敲除技术的基础之上发展了体细胞基因敲除技术[14-15],这是一种新型的研究特定人体基因生理学功能的高效、快速、特异的方法。相比之下,这种体细胞基因敲除技术操作过程更方便,更省时。自90年代起国外已有较多运用体细胞基因敲除技术研究基因结构功能的报道,但是国内尚无相关文献报道[16-20]。本研究试图运用体细胞基因敲除的方法,设计并构建人DNA polβ基因敲除载体,并用此载体转染Fca9706细胞,通过进一步的筛选,建立Fca9706细胞DNA polβ体细胞基因敲除模型,为下一步研究DNA polβ的生物学功能打下牢固的实验基础和依据。
1 材料与方法
1.1 材料限制性核酸内切酶 BglⅡ、HindⅢ、XhoⅠ、ApaⅠ购自日本Takara公司;T4 DNA连接酶购自美国Promega公司;pGFM-T-easy克隆载体购自美国Promega公司;基因组DNA提取试剂盒、RNA提取试剂盒、质粒提取试剂盒、胶回收试剂盒均购自德国QIAGFN公司。食管癌细胞系 Fca9706、大肠杆菌DH5α、克隆载体pcDNA3.1均由本实验室保存。PCR引物由上海生工生物技术服务公司合成。下游同源序列扩增用引物:ND1/ND2为外轮扩增引物,ND3/ND4为内轮扩增引物,ND1 5’-GCCCTGTTCTCTGTAATTTCCACG-3’;ND2 5’-GACCTCAAGCAATTCAGACCCAAGC-3’;ND3 5’-GACAGATCTCCACGAAGCCTTGTGTACTGTGTCAC-3’Bgl II;ND4 5’-AACAAGCTTCCAGAAATGGGCTAGATAGCAACTC-3’HindⅢ。上游同源序列扩增引物:UP1 5’-ACTCGAGGAGAGTGGTCACTGCTCACTG-3’XhoⅠ;UP2 5’-TGGGCCCGCTTGTTGTGACGTCACGCGTC-3’ApaⅠ。pGFM-T Fasy质粒多克隆位点两侧T7、SP6启动子序列设计通用引物:T7 5’-TAATACGACTCACTATAGGGAGA-3’;SP6 5’-CATACGTATTTAGGTGACACTATAG-3’。
1.2 人食管癌Eca9706细胞基因组提取与纯化首先富集扩增培养Fca9706细胞,3~4周后收集细胞。依照德国QIAGFN公司的人基因组DNA提取试剂盒操作说明,提取Fca9706细胞基因组。
1.3 扩增并纯化目的基因上游和下游同源序列根据GenBank中DNA polβ基因序列(M13140),利用UCSC中Blat分析工具软件分析DNA polβ基因的所有内含子、外显子以及启动子序列;选择DNA polβ敲除序列上游和下游同源重组区;设计并合成上游同源重组区(UP)和下游同源重组区(DOWN)PCR引物;以Fca9706细胞基因组 DNA为模板进行扩增。目的DNA通过质量分数1.5%琼脂糖凝胶电泳后,按照快速凝胶回收试剂盒的操作说明,回收该基因片段,此即获得纯化的上游同源序列(UP)。下游同源序列(DOWN)的纯化过程与此相同。
1.4 T-A克隆将上游同源序列(UP)和下游同源序列(DOWN)的纯化产物分别克隆入pGFM-T载体,转化大肠杆菌DH5α株,通过蓝白筛选,挑选白色菌落进行PCR和酶切鉴定,即获得了阳性重组质粒pGFM-TDOWN和pGFM-T-UP。
1.5 构建质粒pOUT-DOWN用小量质粒提取试剂盒提取质粒pcDNA3.1和鉴定正确的pGFM-T-DOWN,经限制性核酸内切酶BglⅡ/HindⅢ双酶切后,分别回收大片段(pcDNA3.1)和小片段(DOWN),具体步骤参照胶回收纯化试剂盒。在T4 DNA连接酶的作用下,室温连接2 h,产生重组质粒pOUT-DOWN。经限制性核酸内切酶BglⅡ/HindⅢ双酶切鉴定后,筛选出鉴定正确细菌,并将其接种于5 mL含有氨苄青霉素(A+)的LB液体培养基中进行扩菌,最后再用质粒提取试剂盒提取构建成功的质粒pOUT-DOWN。
1.6 构建转染用质粒pOUT-polβ提取质粒pOUTDOWN和pGFM-T-UP,分别用XhoⅠ/ApaⅠ双酶切,获得线性化的pOUT-DOWN和小片段(UP)用凝胶回收试剂盒进行纯化。纯化产物再用T4 DNA连接酶进行连接,室温连接2 h,将连接产物转化入感受态细胞大肠杆菌DH5α中,再在含氨苄青霉素的LB平板上随机挑取5个单菌落,通过酶切和PCR扩增,筛选得到基因敲除质粒pOUT-polβ。基因敲除载体结构示意图见图1。
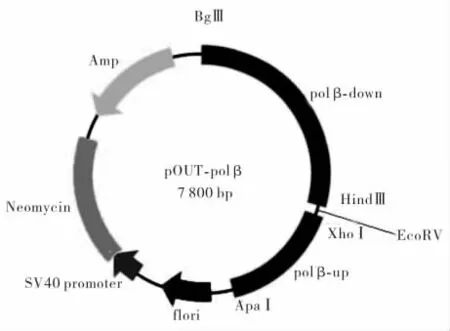
图1 基因敲除载体pOUT-polβ结构示意图
2 结果
2.1 基因敲除序列UP区段和DOWN区段扩增结果
PCR扩增Fca9706细胞基因组DNA,分别得到UP和DOWN同源序列。下游同源序列DOWN扩增产物,长度约为1 756 bp(图2);上游同源序列UP扩增产物,长度约为1 280 bp(图3)。
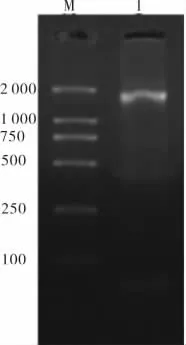
图2 DNA polβ下游同源序列DOWN的扩增结果
2.2 上游和下游基因敲除同源序列与pGEM-T载体克隆重组子鉴定结果以T7/SP6为引物,PCR扩增鉴定重组载体pGFM-T-UP,pGFM-T-DOWN,分别得到约为1 444 bp(UP)和1 932 bp(DOWN)的阳性克隆扩增片段。见图4、5。

图3 DNA polβ上游同源序列的扩增结果

图4 重组质粒pGFM-T-up,扩增产物电泳结果

图5 重组质粒pGFM-T-down扩增产物电泳结果
2.3 骨架载体pcDNA3.1与下游同源序列DOWN重组结果用限制性核酸酶切酶BglⅡ/HindⅢ双酶切该重组载体,电泳可见,在同一泳道上有2条明亮条带分别位于1 756 bp处和3 622 bp处,与设计一致。见图6。
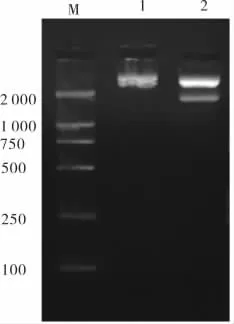
图6 骨架载体pcDNA3.1与下游同源序列DOWN重组结果
2.4 基因敲除重组载体pOUT-polβ鉴定结果用引物UP1/UP2,PCR扩增鉴定;限制性核酸内切酶XhoⅠ/ApaⅠ、BglⅡ/HindⅢ双酶切鉴定,FcoR V单酶切鉴定,验证下游和上游同源序列是否正确插入到骨架载体中。电泳结果与设计一致。见图7、8。
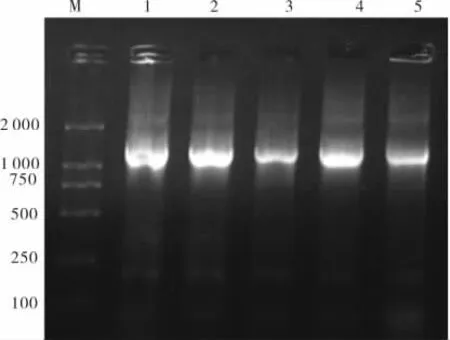
图7 引物UP1/UP2扩增鉴定结果
2.5 线性化基因敲除载体鉴定结果提取鉴定正确的基因敲除重组载体,经FcoR V酶切使其线性化,电泳结果显示,有一条明亮条带位于5 428 bp处。见图9。
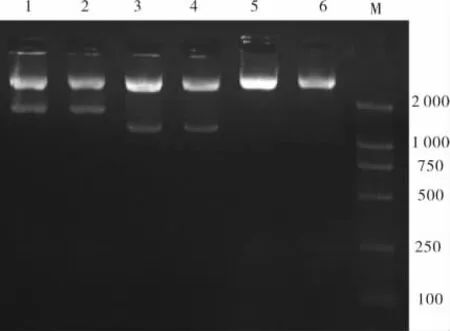
图8 酶切鉴定重组质粒电泳结果

图9 线性化基因打靶载体经酶切鉴定结果
3 讨论
随着后基因组时代的到来,基因研究的热点开始转向基因及其表达产物的生物学功能。通过构建细胞模型,采用目的基因过量表达和(或)反义抑制等方法来研究基因的生理学功能。但反义抑制不能完全消除细胞内源基因的表达作用,往往对结果分析造成一定干扰。基因敲除通过同源重组,将细胞特定基因破坏,从而造成该基因功能丧失,能够完全消除细胞内源基因的干扰。体细胞基因敲除技术,由于其去除了动物胚胎操作以及交配繁殖生长等过程,从而大大缩短时间,因此,体细胞基因敲除技术已成为一种研究特定人体基因生理学功能的高效快速特异的方法。
由于同源重组的过程中存在着重组效率低,以及目的片段的随机插入等诸多因素的影响,所以,基因敲除载体的设计和构建以及筛选系统的选择将直接影响到基因敲除策略和效率[21],并且决定着整个实验结果的成败。本研究所构建的基因敲除载体全长约7.8 kb,基因结构为-5’上游同源序列-neo-下游同源序列-3’-,其中上游同源序列长约1 268 bp,下游同源序列长约2 150 bp,包含了DNA polβ基因第1、2、3外显子和第1、2内含子的全部以及第3内含子的大部分,并且还包含了DNA polβ基因启动子的一部分。选用neo基因作为选择标志基因,利用含有正筛选基因neo来提高同源重组率,若同源重组如期发生,基因敲除载体的同源序列将取代与DNA polβ基因相应的同源区,而且,由于DNA polβ基因外显子的一部分也在同源重组的过程中被敲除[22-23],这样将导致DNA polβ基因功能的丧失。构建成功基因敲除载体通过限制性核酸内切酶切的图谱分析及测序证实,符合预期设计的结构。将此基因敲除载体导入受体细胞,与靶基因发生同源重组。通过G-418筛选,最终获得DNA polβ基因缺失的细胞模型,这为后续研究人食管癌DNA polβ的生物学功能工作打下实验基础和依据。
[1] Srivastava DK,Husain I,Arteaga CL,et al.DNA polymerase beta expression differences in selected human tumors and cell lines[J]. Carcinogenesis,1999,20(6):1049-1054.
[2] 任昀.DNA聚合酶β和肿瘤[J].癌变·畸变·突变,2004,16(2):125-128.
[3] Bhattacharyya N,Chen HC,Grundfest-Broniatowski S,et al.Alteration of hMSH2 and DNA polymerase beta genes in breast carcinomas and fibroadenomas[J].Biochem Biophys Res Commun,1999,259(2):429-435.
[4] Iwanaga A,Ouchida M,Miyazaki K,et al.Functional mutation of DNA polymerase beta found in human gastric cancer--inability of the base excision repair in vitro[J].Mutat Res,1999,435(2):121 -128.
[5] Fydmann MF,Knowles MA.Mutation analysis of 8p genes POLB and PPP2CB in bladder cancer[J].Cancer Genet Cytogenet,1997,93(2):167-171.
[6] Bergerheim US,Kunimi K,Collins VP,et al.Deletion mapping of chromosomes 8,10,and 16 in human prostatic carcinoma[J]. Genes Chromosomes Cancer,1991,3(3):215-220.
[7] Dobashi Y,Shuin T,Tsuruga H,et al.DNA polymerase beta gene mutation in human prostate cancer[J].Cancer Res,1994,54(11):2827-2829.
[8] Hübscher U,Nasheuer HP,Syväoja JF.Fukaryotic DNA polymerases,a growing family[J].Trends Biochem Sci,2000,25(3):143-147.
[9] Iwanaga A,Ouchida M,Miyazaki K,et al.Functional mutation of DNA polymerase beta found in human gastric cancer--inability of the base excision repairin vitro[J].Mutat Res,1999,435(2):121 -128.
[10]董子明,赵国强,赵勤,等.人食管癌组织中DNA聚合酶β基因突变的研究[J].中华医学杂志,2002,82(13):899-902.
[11]Srivastava DK,Husain I,Arteaga CL,et al.DNA polymerase beta expression differences in selected human tumors and cell lines[J]. Carcinogenesis,1999,20(6):1049-1054.
[12]Capecchi MR.Altering the genome by homologous recombination[J].Science,1989,244(4910):1288-1292.
[13]董子明.DNA聚合酶β的研究现状[J].郑州大学学报:医学版,2003,38(4):477-479.
[14]Sedivy JM,Dutriaux A.Gene targeting and somatic cell genetics--a rebirth or a coming of age?[J].Trends Genet,1999,15(3):88-90.
[15]Sedivy JM.Gene targeting comes to top-down drug screens[J]. Trends Biotechnol,2002,20(3):92-93.
[16]Brown JP,Wei W,Sedivy JM.Bypass of senescence after disruption of p21CIP1/WAF1 gene in normal diploid human fibroblasts[J]. Science,1997,277(5327):831-834.
[17]Chan TA,Hermeking H,Lengauer C,et al.14-3-3Sigma is required to prevent mitotic catastrophe after DNA damage[J].Nature,1999,401(6753):616-620.
[18]Park BH,Vogelstein B,Kinzler KW.Geneticd isruption of PPARdelta decreases the tumorigenicity of human colon cancer cells[J].Proc Natl Acad Sci U S A,2001,98(5):2598-2603.
[19]Li G,Nelsen C,Hendrickson FA.Ku86 is essential in human somatic cells[J].Proc Natl Acad Sci U S A,2002,99(2):832 -837.
[20]Shulman MJ,Nissen L,Collins C.Homologous recombination in hybridoma cells:dependence on time and fragment length[J].Mol Cell Biol,1990,10(9):4466-4472.
[21]Thomas KR,Capecchi MR.Site-directed mutagenesis by gene targeting in mouse embryo-derived stem cells[J].Cell,1987,51(3):503-512.
[22]生秀杰.基因打靶的策略及其发展[J].国外医学:遗传学分册,2001,24(1):8-10.
[23]Zhang Y,Yuan F,Wu X,et al.Preferential incorporation of G opposite template T by the low-fidelity human DNA polymerase iota[J].Mol Cell Biol,2000,20(19):7099-7108.
Construction of Eca9706 Cell DNA Polβ Gene Targeting Vector
Feng Long1,2,Guo Wentao3,Lu Wuhao4,Sun Sajia2,Dong Ziming2
(1.College of Basic Medical Science,Henan University of TCM,Zhengzhou 450008,China;2.College of Basic Medical Science,Zhengzhou University,Zhengzhou 450001,China;3.Luohe Medical college,Luohe 462002,China;4.The First Affiliated Hospital,Zhengzhou University,Zhengzhou 450052,China)
ObjectiveTo construct Fca9706 cell DNA polymerase β(DNA polβ)gene targeting vector.Methods According to the principle of somatic gene targeting and DNA polβ sequence,the two special primers were designed and synthesized(UP1/UP2 and DOWN1/DOWN2)to amplify the up homologous sequence and the down homologous sequence.The up sequence(1 268 bp)and the down sequence(2 150 bp)were inserted into skeleton vector pcDNA3.1,then DNA polβ gene targeting vector pOUT-polβ was constructed.Finally,the targeting vector was identified by PCR,restriction enzyme digestion and sequencing.ResultsThe up sequence and the down sequence were exactly inserted into skeleton vector pcDNA3.1,then DNA polβ gene targeting vector pOUT-polβ was constructed.ConclusionThe Fca9706 cell DNA polβ gene targeting vector was established successfully by using the method in this thesis.
gene targeting;DNA polymerase;targeting vector;Fca9706 cell
10.3969/j.issn.1673-5412.2014.03.001
R735.1;R730.23
A
1673-5412(2014)03-0185-05
2014-03-14)
国家自然科学基金资助项目(编号:30471952)
冯龙(1981-),女,博士,讲师,主要从事肿瘤发病及病因学研究。F-mail:flong01@163.com
董子明(1953-),教授,博士生导师,主要从事肿瘤病理生理学相关研究。F-mail:dongzmzzu@126.com
猜你喜欢
——一道江苏高考题的奥秘解读和拓展
中学生物学(2022年7期)2022-09-07
成都医学院学报(2022年4期)2022-08-19
基层中医药(2022年4期)2022-07-22
汉字汉语研究(2021年2期)2021-08-30
江西农业学报(2021年4期)2021-04-20
三农资讯半月报(2020年11期)2020-06-21
汉字汉语研究(2019年2期)2019-08-27
天津医科大学学报(2019年3期)2019-08-13
新高考·英语进阶(高二高三)(2018年8期)2018-01-15
腹腔镜外科杂志(2016年9期)2016-06-01
