
多发性神经纤维瘤病合并颈部恶性神经鞘膜瘤1例
2014-06-01 09:24张宇鹏张建刚包海军丁茂盛
中国癌症杂志 2014年9期
张宇鹏张建刚包海军丁茂盛
1.宁夏医科大学临床医学院,宁夏 银川 750004;
2.甘肃省第二人民医院胸外科,甘肃 兰州 730000;
3.兰州大学病理学研究所,甘肃 兰州730000
多发性神经纤维瘤病合并颈部恶性神经鞘膜瘤1例
张宇鹏1张建刚2包海军3丁茂盛3
1.宁夏医科大学临床医学院,宁夏 银川 750004;
2.甘肃省第二人民医院胸外科,甘肃 兰州 730000;
3.兰州大学病理学研究所,甘肃 兰州730000
多发性神经纤维瘤;恶性神经鞘瘤;诊断;治疗
多发性神经纤维瘤病(neurofibromatosis,NF)也称冯雷克林霍增氏病,为常染色体显性遗传疾病,发病率为1/3300~1/2500[1],系外胚层和中胚层组织发生障碍所致,多属斑痣性错构瘤,而非真性肿瘤。恶性周围神经鞘膜瘤(malignant peripheral nerve sheath tumor,MPNST)或称恶性雪旺氏瘤、神经源性肉瘤、神经纤维肉瘤,是一种具有神经分化潜能的恶性软组织肿瘤,约占全部软组织肉瘤的5%。MPNST起源于外胚层间质,由纤维母细胞、EMA阳性的束膜细胞、雪旺细胞、CD34阳性的树突状细胞、原始上皮细胞等构成的一个异质性肿瘤。此肿瘤呈高度恶性,常发生于神经干,以头颈部多见,其次是躯干、四肢等部位,后腹膜、盆腔等处少见,还有报道发生于肝脏的恶性外周神经鞘膜瘤[2],存活率较低。MPNST的发病率大约是0.000 1%,其中4.6%的患者合并多发性神经纤维瘤病Ⅰ型(NF1)[3],研究表明有NF1病史的患者患MPNST的风险是正常人的18倍[4]。本文报道1例NF1合并恶性神经鞘膜瘤的病例,结合文献资料对其临床表现、病理及免疫组化探讨如下。
1 患者资料
患者男性,61岁,以全身体表多发串珠样结节30年,右侧颈部肿块伴疼痛8年于2014年1月14日入院。近8年已行4次颈部肿物切除术,均复发。查体:神志清醒,精神较好,心肺腹未见异常。头部、颈项部、躯干、四肢皮肤大量串珠状瘤样结节(图1),无压痛,直径0.3~4.0 cm,大小不等,部分高出皮肤1.0~2.0 cm;右侧颈项部可见一巨大肿物,大小约15.0 cm×8.0 cm×6.0 cm实质性肿块(图2),呈分叶状,质硬,移动度小,有触痛,皮肤表面见1处2.0 cm×2.0 cm溃疡,周边皮肤有溃烂,溃烂区为2.5 cm× 1.5 cm。颈部CT显示:项部软组织不对称,右侧皮下见较大不规则形软组织密度影,CT值约40 Hu,最大层面大小为120.0 mm×70.0 mm×69.0 mm,边界尚清,有明显分叶,形态不规则,邻近软组织明显受压,病变外缘局部见絮状较高密度影:余颈项部皮下及皮肤表面可见多个小结节样软组织样密度影凸起(图3)。结合以上检查结果及病史,临床诊断为:①NF1;②右颈部肿物性质待查。询问患者家族史,未发现其他人罹患此病。
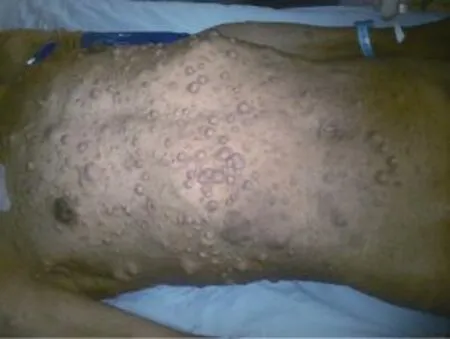
图1 典型牛奶咖啡样色斑及大小不等结节Fig. 1 A typical cappuccino-like spots and nodules of varying sizes of the patient
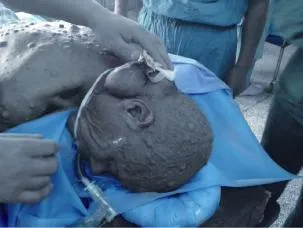
图2 患者颈部巨大肿瘤Fig. 2 Clinical appearance of the tumor
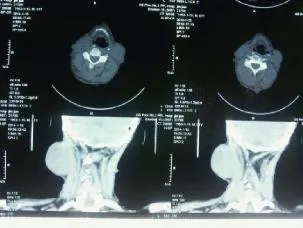
图3 颈部磁共振显像Fig. 3 Neck magnetic resonance imaging
患者于2014年1月17日在全麻下行右颈部肿块切除术。取右侧肿物与正常皮肤组织交界处,逐层切开皮肤及皮下组织,术中见肿瘤上界达颅底,下界达斜方肌上缘,前界达胸锁乳突肌后缘,与颅骨及胸锁乳突肌粘连,常规游离并切除肿块及部分正常组织,瘤体约15.0 cm×8.0 cm×5.0 cm,无包膜。病理所见:肉眼观肿瘤呈灰白灰红色结节状,大小14.0 cm×8.5 cm× 6.0 cm,上带部分皮肤组织,皮肤面积为8.0 cm×5.5 cm,皮面上可见2.0 cm×2.0 cm溃疡,底部污秽,周围略隆起,周边皮肤有溃烂,溃烂区大小约为2.5 cm× 1.5 cm(图4)。结节切面灰白灰黄色,局部鱼肉状。光镜所见:肿瘤组织,皮肤溃疡形成,肿瘤细胞束状、交织状排列,局部见交替性分布的细胞丰富区和细胞稀疏区域,瘤细胞核不规则,部分核异型性明显,核分裂像多见,多灶坏死,横纹肌束间肿瘤组织。病理诊断为(右项部)恶性周围神经鞘膜瘤(图5)。免疫组化:瘤细胞SMA (-);Desmin(-);Myoglobin(-);S-100(+++)(图6);CD117(-);CD34(-);Vimentin(+、++);CKP(-); MyoD1(-);Bcl-2(-);MSA(-);EMA(±);NF(±);P53 (-);Ki-67(+,15%~20%)。
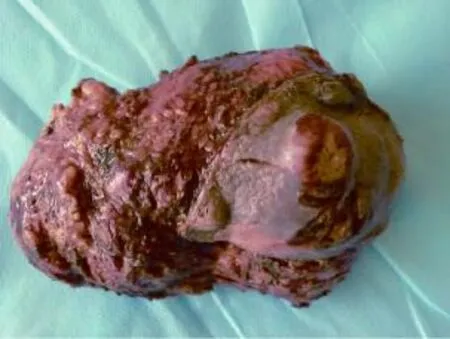
图4 颈部巨大肿物Fig. 4 The grossary appearance of the huge tumor

图5 颈部肿物切片Fig. 5 Microscopic appearance of the tumor(HE, ×200)
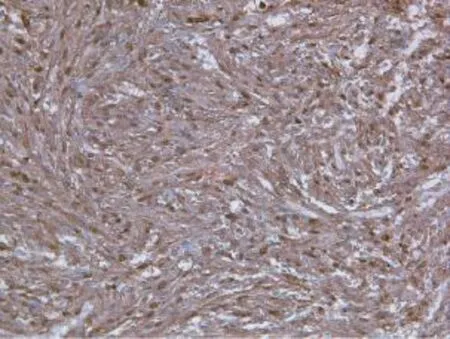
图6 肿瘤组织免疫组化S-100 (+++)Fig. 6 Diffuse S-100 positivity detected by immunohistochemistry (+++)(×200)
2 讨 论
NF1临床上具有五大特点:①多发性皮肤结节和瘤块;②皮肤上出现牛奶咖啡样色斑,常在儿童或青年期先于肿瘤出现;③周围神经干串珠状神经纤维瘤样增粗;④局部神经纤维瘤性象皮病;⑤伴有其他畸形、呆滞及黑色素瘤。因其临床表现呈多态性,Riecardi于1982年将本病区分为8个类型:Ⅰ型即经典型,占85%~90%,目前认为NF1发病与常染色体17q11.2上抑癌基因Nf1突变有关。Nf1的基因编码产物称为神经纤维蛋白,具有抑制肿瘤的作用,其作用之一是通过灭活癌基因p21-ras,而阻断其细胞内信号通路。NF1以神经纤维瘤、牛奶咖啡样色斑、骨质畸形、智力障碍和虹膜错构瘤为特征;Ⅱ型为中央型,表现为双侧听神经瘤伴少量色斑;Ⅲ型为兼有皮肤多发性结节与中枢神经系统肿瘤;Ⅳ型为伴有色斑的弥漫性神经性肿瘤;Ⅴ型为局限性的常有色斑的神经纤维瘤;Ⅵ型则只有色斑,此型多不具遗传性;Ⅶ型为迟发性,多在20多岁以后出现异常;上述之外的则为Ⅷ型。本病瘤样结节生长缓慢,有时可自动停止,但国外报道有10%~15%的恶变率,多数恶变为神经纤维肉瘤、恶性神经鞘瘤,偶尔恶变为横纹肌肉瘤、血管肉瘤等[5]。1987年美国国立卫生研究院(National Institute of Health,NIH)提议,将NF分为NF1型和NF2型,即周围型和中枢型神经纤维瘤病,两者主要区别是有不同的突变基因和临床特征,其中NF1较常见;而NF2主要指颅内神经纤维瘤病,发病率约为1∶1 000 000,多在20~40岁之间。NF2的特征性为双侧听神经瘤,表现为听力逐渐下降,同时伴有耳鸣、头痛、平衡失调等症状。NF2型的诊断和治疗相对困难,预后较差。MPNST旧称很多,分为3种:①恶性神经瘤,由神经鞘细胞发生;②恶性神经纤维瘤,由神经鞘、神经纤维和神经束衣、内衣发生;③神经鞘纤维肉瘤,由神经内衣母细胞或神经束衣发生。为避免混淆,WHO于1994年将上述名称正式采纳为恶性外周神经鞘膜瘤,包括了来源于外周神经、神经纤维瘤恶变或呈神经鞘膜分化的梭形细胞肉瘤等多个肿瘤。MPNST可发生在周围神经、颅神经和交感神经,好发于20~50岁,多发性神经纤维瘤恶变的MPNST有17号染色丢失及p53基因突变。
NF1与MPNST关系密切,约有50%的MPNST来源于NF1[4]。NF1合并MPNST最早由挪威Oslo大学的病理学家Harbitz[6]报道了2例,目前为止,除部分地区大型医院有较大样本量的报道外[1,7-11],多数文献仍以个案报道为主[3,12-14]。一项针对MPNST生存的Meta分析从1970—2011年也仅收集到62例NF1合并MPNST的资料(>5例的研究)[15]。查阅相关文献,国内目前报道的NF合并颈部神经鞘瘤的个案报道较少[16-20]。神经鞘瘤恶变极为罕见,结合本例,患者颈项部恶性神经鞘膜瘤或许与8年来反复不规范手术切除肿物及切除后患者反复搔抓或衣物摩擦刺激肿物有关。MPNST是一种形态学瘤谱范围广、免疫表型异质性大的恶性肿瘤,由于缺乏标准的诊断模式,目前仍然是软组织肿瘤中最难诊断的肿瘤之一。免疫标志物有助于诊断,SMA(HHF35)是区别MPNST与平滑肌肉瘤有价值的指标。S100为MPNST最敏感的标志物,髓脂碱性蛋白为最可靠的标志物。总体表现为Vimentin、S100、EMA、CK阳性,SMA阴性,但同一肿瘤及不同肿瘤间其阳性区域、分布特点、上述抗原表达的数目及类型会呈现明显不同。MPNST的免疫表型呈现明显的异质性,提示在对MPNST进行免疫标志物检测时,应结合多种指标,以防漏诊或误诊。NF1目前尚无特异治疗,如无恶变,除影响外观外,可长期带瘤生存,对生命不构成威胁,但应长期观察。倘若伴发恶性神经鞘瘤则首选以手术为主包括放化疗的综合治疗。如为低度恶性,及早合理的治疗,尚可获得一定的远期存活率。目前治疗MPNST采取以手术切除肿块为主、化疗及放疗为辅的综合治疗,手术应尽可能广泛地切除病变及周围组织,如果发生在肢体,必要时应截肢,这是治疗成功的关键,术后辅助放疗可控制或延迟局部病灶复发,与其他良性肿瘤相比,MPNST术后易复发,并扩展到邻近组织,多次手术易发生肺、脑等远处转移,晚期可发生淋巴道及血行转移,也可转移至胸、腹腔等。大多数MPNST是高度恶性肿瘤,生长速度相对缓慢,侵袭力较弱,合并神经纤维瘤的MPNST患者预后差,5年生存率为16%~38%[21]。尽管放疗对提高长期存活率作用不大,但仍属MPNST的有效治疗手段之一,而对肿瘤组织无法根治性切除的患者应选姑息性切除+大剂量放疗,尤其是早期放疗对提高临床缓解率有重要作用。术后化疗可防止肿瘤远处转移,但有效的药物很少,单一用药常选择多柔比星,亦可选用多柔比星和异环磷酰胺联合用药。另外研究发现,输注免疫球蛋白可能是MPNST辅助治疗的有效方法,有助于延长MPNST患者的无瘤生存时间。
[1] OUIDAD Z, ELIZABETH F, LAURENT Z, et al. Chemotherapy for the treatment of malignant peripheral nerve sheath tumors in neurofibromatosis 1:a 10-year institutional review [J]. Orphanet J Rare Dis, 2013, 8: 127.
[2] KÓBORI L, NAGY P, MÁTHÉ Z, et al. Malignant peripheral nerve sheath tumor of the liver: a case report [J]. Pathol Oncol Res, 2008, 14(3): 329-332.
[3] HSU C C, HUANG T W, HSU J Y, et al. Malignant peripheral nerve sheath tumor of the chest wall associated with neurofibromatosis: a case report [J]. J Thorac Dis, 2013, 5(3): 78-82.
[4] TUCKER T, WOLKENSTEIN P, REVUZ J, et al. Association between benign and malignant peripheral nerve sheath tumors in NF1 [J]. Neurology, 2005, 65(2): 205-211.
[5] 黄方. 多发性神经纤维瘤病伴颈部恶性神经鞘瘤一家系[J]. 癌症, 2000, 19(5): 505.
[6] HARBITZ F. Multiple neuro fi bromatosis (von Recklinghausen’s disease) [J]. Arch Int Med, 1909, 3: 32-65.
[7] LAFEMINA J, QIN L X, MORACO N H, et al. Oncologic outcomes of sporadic, neurofibromatosis-associated, and radiation-induced malignant peripheral nerve sheath tumors[J]. Ann Surg Oncol, 2013, 20(1): 66-72.
[8] LEROY K, DUMAS V, MARTIN-GARCIA N, et al. Malignant peripheral nerve sheath tumors associated with neurofibromatosis type 1: a clinicopathologic and molecular study of 17 patients [J]. Arch Dermatol, 2001, 137(7): 908-913.
[9] EVANS D G, BASER M E, MCGAUGHRAN J, et al. Malignant peripheral nerve sheath tumours in neurofibromatosis 1 [J]. J Med Genet, 2002, 39(5): 311-314.
[10] DUNN G P, SPILIOPOULOS K, PLOTKIN S R, et al. Role of resection of malignant peripheral nerve sheath tumors in patients with neurofibromatosis type 1 [J]. J Neurosurg, 2013, 118(1): 142-148.
[11] KAMRAN S C, HOWARD S A, SHINAGARE A B, et al. Malignant peripheral nerve sheath tumors: prognostic impact of rhabdomyoblastic differentiation (malignant triton tumors), neurofibromatosis 1 status and location [J]. Eur J Surg Oncol, 2013, 39(1): 46-52.
[12] LEE D S, JUNG S I, KIM D W, et al. Malignant peripheral nerve sheath tumor arising from neurofibromatosis [J]. Arch Plast Surg, 2013, 40(3): 272-275.
[13] GALATIAN A A, CROWSON A N, FISCHER R J, et al. Malignant peripheral nerve sheath tumor with glandular differentiation in a patient with neurofibromatosis type 1[J]. Am J Dermatopathol, 2013, 35(8): 859-863.
[14] ENDO M, YAMAMOTO H, HARIMAYA K, et al. Conventional spindle cell-type malignant peripheral nerve sheath tumor arising in a sporadic schwannoma [J]. Hum Pathol, 2013, 44(12): 2845-2848.
[15] KOLBERG M, HØLAND M, AGESEN T H, et al. Survival meta-analyses for >1800 malignant peripheral nerve sheath tumor patients with and without neurofibromatosis type 1[J]. Neuro Oncol, 2013, 15(2): 135-147.
[16] 瞿文瑞. 神经纤维瘤病的临床分析(附16例报告) [D].吉林大学, 2012.
[17] 黄方, 吴春林. 多发性神经纤维瘤病伴颈椎椎管内外沟通性神经鞘瘤1例 [J]. 罕少疾病杂志, 2004, 11(1): 41-42.
[18] 滕荣跃. 恶性外周神经鞘瘤10例临床分析 [D]. 浙江大学, 2009.
[19] 方黎明, 吕雄根. 多发性神经纤维瘤病一家系调查与染色体变化 [J]. 浙江医学, 1995, 17(5): 319.
[20] WENG Y, CHEN Y, CHEN J, et al. Common genetic variants in the microRNA biogenesis pathway are associated with malignant peripheral nerve sheath tumor risk in a Chinese population [J]. Cancer Epidemiol, 2013, 37(6): 913-916.
[21] PORTER D E, PRASAD V, FOSTER L, et al. Surbibal in malignant peripheral nerve sheath tumours: a comparison between sporadic and neurofibromatosis type 1-Associated tumours [J]. Sarcoma, 2009, 2009: 75639.
10.3969/j.issn.1007-3969.2014.09.013
R739.43
A
1007-3639(2014)09-0717-04
2014-03-06
2014-04-28)
张宇鹏 E-mail:zhangyp149@sohu.com
猜你喜欢
中国临床医学影像杂志(2022年2期)2022-05-25
天津医科大学学报(2021年3期)2021-07-21
中华养生保健(2020年7期)2020-11-16
中国临床医学影像杂志(2019年6期)2019-08-27
中国临床医学影像杂志(2019年4期)2019-06-18
中国临床医学影像杂志(2019年4期)2019-06-18
中国现代神经疾病杂志(2018年2期)2018-05-09
中国医疗美容(2015年1期)2015-07-12
郑州大学学报(医学版)(2015年1期)2015-02-27
中国医药指南(2013年23期)2013-01-23
