
基于procaspase-3激活的抗肿瘤活性分子SM-1吸收机制研究
2014-05-14 11:21罗丽娜张海龙郑志难丁劲松
中国药理学通报 2014年4期
唐 靖,罗丽娜,张海龙,郑志难,张 杰,李 溯,何 花,丁劲松

Fig 1 Chemical structure of PAC-1 and SM-1
SM-1能特异性激活肿瘤细胞凋亡信号转导通路关键酶原半胱天冬蛋白酶-3酶原(procaspase-3),活化为肿瘤凋亡关键执行分子半胱天冬酶3(caspase-3),而导致肿瘤细胞快速死亡[2]。前期研究发现,其对主要肺癌细胞株(如NCI-H226、NCIH460)的抑制活性超过 PAC-1[3-4](目前唯一进入Ⅱ期临床的同作用机制的药物)约8~10倍,对裸鼠A549细胞株移植瘤的抑瘤率较后者强50%,且毒性更低。基于初步药效学、作用机制、药动学特征及毒性等研究,SM-1有望成为一种新型的抗肿瘤药物,特别是针对非小细胞肺癌。
本文在SM-1基本理化性质研究的基础上(如溶解度、亲脂性、稳定性等),采用 Caco-2细胞单层模型[5]及大鼠在体单向肠灌流模型[6]研究 SM-1的吸收特征,考察化合物浓度对吸收的影响,研究其有效肠吸收部位,以及吸收过程是否受转运蛋白外排作用影响。通过SM-1在大鼠体内绝对生物利用度研究,初步探讨其药代动力学特点,综合评价 SM-1在体内的吸收程度,为 SM-1的成药性评价以及剂型设计提供理论依据。
1 材料与方法
1.1 仪器与试药 LC-20AT高效液相色谱仪(日本岛津公司);细胞电位仪(World Precision Instruments Inc);Beckman coulter DTX880酶标仪(美国 Beckman Coulter公司);Transwell板(0.2μm pore diameter,12 mm diameter,美国 Corning公司);BT100-1F蠕动泵(保定兰格恒流泵有限公司)。
SM-1(深圳市湘雅生物医药研究院提供,含量:99.7%,批号:M2K142);Caco-2细胞株(湘雅医院细胞库);RPMI 1640培养基(美国HyClone公司);胎牛血清(天津市灏洋生物制品科技有限公司);非必需氨基酸、0.25%胰蛋白酶-EDTA、Hank’s缓冲液(美国Gibco公司),其他试剂均为分析纯。
1.2 细胞培养与毒性实验 Caco-2细胞按参考文献[7]培养约21 d后,细胞形成紧密的单层,采用跨上皮细胞电阻和普萘洛尔渗透性实验检测细胞的完整性与渗透性。并以MTT法测定SM-1对Caco-2细胞的毒性,以安全浓度范围的SM-1进行转运实验。
1.3 Caco-2单层细胞模型双向转运实验 AP→BL侧的转运实验中,在 AP侧加入10、20、40 mg·L-1SM-1 HBSS溶液0.5 ml,BL侧加入空白HBSS 1.5 ml。BL→AP侧的转运实验中,在 BL侧加入10、20、40 mg·L-1SM-1 HBSS溶液1.5 ml,AP侧加入空白HBSS 0.5 ml。置37℃孵育,持续振摇(50 r·min-1),分别于 30、60、90、120、180 min收集 A侧或B侧溶液100μl,每次取样后补足相同体积的HBSS溶液至Transwell中。
1.4 大鼠在体肠灌流实验 健康♂SD大鼠(禁食12 h),200~220 g,SPF级(湖南斯莱克景达实验动物有限公司)。腹腔注射体积分数为0.1的水合氯醛(0.04μl·g-1)麻醉后,手术过程、药物灌流方法同参考文献[8]。选择空肠段进行灌流,分别考察100、50、25 mg·L-1浓度下 SM-1在不同肠段的吸收情况。采用重量法标示灌流液体积。
[10]Anderson,L.M.,&Pearson,C.M.:Tit for Tat The Spiraling Effects of Incivility in the Workplace.Academy of Management Review,1999,24.
1.5 大鼠体内绝对生物利用度研究 12只♂SD大鼠随机分为2组(禁食12 h),一组口服灌胃给药100μg·g-1的 SM-1(0.5%吐温-80混悬液),于给药后 0.25、0.5、1、1.5、2、3、4、6、8 h眼眶取血;一组静脉注射12.5μg·g-1的SM-1(体积分数为0.1的丙二醇生理盐水溶液),于给药后0.08、0.33、0.67、1、1.5、2、3、4、6 h取血。每次取血 0.5 ml置于肝素化离心管,5 000×g离心10 min,分离血浆,置于-20℃冰箱保存待测。
1.6 样品分析与计算 色谱柱:Diamonsil C18(250 mm×4.6 mm,0.5μm);流动相:甲醇 -0.01 mol·L-1(pH 6.8)PBS(87∶13);流速:1.0 ml·min-1;检测波长:325 nm;进样量:50μl。
样品(血浆、肠灌流液)均与甲醇以1∶2处理,涡旋混匀,离心(80 000×g,4℃,10 min),取上清液进样分析。分别考察SM-1 HBSS溶液在细胞转运实验过程中的稳定性,SM-1肠灌流液在37℃水浴2 h的稳定性,硅胶管路及肠壁对SM-1肠灌流液的吸附性。
1.6.1 双向转运实验 表观渗透系数(Papp)根据以下公式计算。
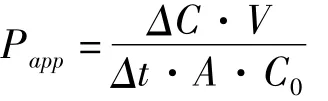
公式中,C0为给药侧药物初始浓度,ΔC/Δt为接收侧药物出现的速率,V为接收侧体积,A为Transwell多聚碳酸酯膜表面积。
1.6.2 在体肠吸收实验 吸收速率常数(Ka)和肠有效渗透系数(Peff)分别采用以下公式计算。
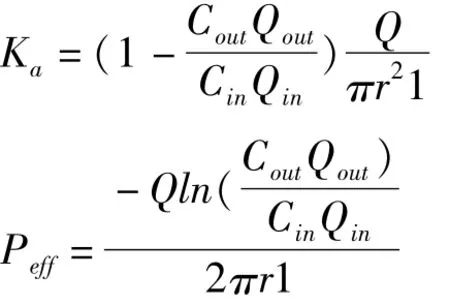
公式中,Cin和Cout为肠道进、出口的灌流液中SM-1浓度,Qin和 Qout为肠道进、出口的灌流液体积(ml)(根据测得重量计算体积,灌流液的密度经校正计算),l和r为灌流肠段长度(cm)与横截面半径 (cm),Q为灌流的速度 (0.2 ml·min-1)。Ka和Peff为30~105 min时间段样品的平均值。
1.6.3 绝对生物利用度 大鼠血药浓度数据采用DAS 2.1药动学应用程序处理,绝对生物利用度Fabs/%采用以下公式计算。
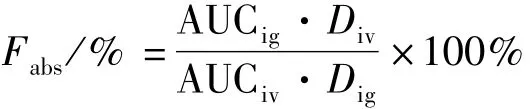
公式中,AUCig为灌胃给药后药时曲线下面积,AUCiv为静脉给药后药时曲线下面积,Dig为灌胃给药剂量,Div为静脉给药剂量。
1.7 统计学处理 实验数据以¯x±s表示,运用SPSS 13.0进行方差分析,比较各组变量间有无差异。
2 结果
2.1 样品测定方法学结果 HBSS溶液中SM-1的标准曲线方程为:Y=17211.4C-589.1(R=0.9998),线性范围:0.1~50 mg·L-1;肠灌流液中SM-1的标准曲线方程为:Y=16922.9C-674.3(R=0.9995),线性范围:0.5~100 mg·L-1;血浆中SM-1的标准曲线方程为:Y=6692.5C-262.4(R=0.9998),线性范围:0.2~20 mg·L-1。SM-1的最低检测限为0.0125 mg·L-1。SM-1保留时间为7.3 min。本实验所采用的色谱条件下,SM-1峰形良好,无杂峰干扰测定,方法的日内精密度、日间精密度、冻融试验结果均符合生物样品分析的一般要求。
低、中、高浓度SM-1的HBSS和灌流液于37℃放置,以及灌流液于37℃循环并与肠壁共孵育后,其含量变化在94.8%~103.31%之间。因此,可认为SM-1在细胞转运实验和肠灌流实验过程中稳定性良好。
2.2 细胞培养和毒性实验 细胞模型的TEER>300Ω·cm2,跨细胞被动转运标记物普萘洛尔表观渗透系数(Papp)为(29.39±0.30)×10-6cm·s-1,与文献[6]报道的 27.5×10-6cm·s-1接近,结果提示该模型的完整性与渗透性均符合要求,可作为研究小肠吸收的模型。当 SM-1浓度≤40 mg·L-1时,OD值与对照组比较差异无显著性(P>0.05),因此采用安全浓度范围内的10、20、40 mg·L-1进行双向转运实验。
2.3 Caco-2细胞模型中 SM-1双向转运实验,不同浓度SM-1在3 h内的累积吸收量及分泌量见Tab 1,其透过量与浓度呈线性关系(R分别为0.965、0.940)。 不 同 浓 度 SM-1的 Papp(AP→BL)、Papp(BL→AP)和 PDR(Papp(BL→AP)/Papp(AP→BL))见 Tab 2。方差分析显示Papp在剂量与方向间差异均无显著性(P>0.05),PDR值约为1。因此,在10~40 mg·L-1时SM-1的吸收以被动扩散为主,其双向转运过程可能与浓度无关,且不受外排转运蛋白的影响。
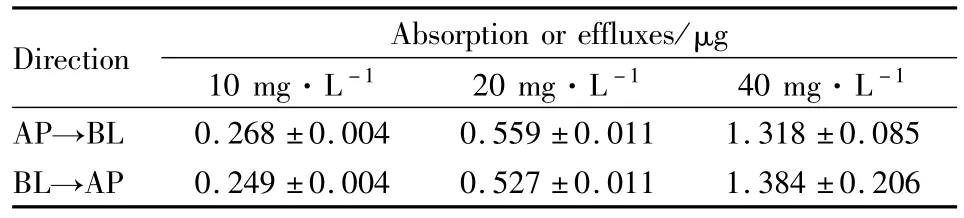
Tab 1 Cumulative absorption and effluxes of SM-1 with different concentrations(¯x±s,n=3)
2.4 在体肠吸收实验 不同浓度药物的吸收参数见 Tab 3。在25~100 mg·L-1内,SM-1的Ka与Peff差异无显著性(P>0.05),说明药物吸收无自身浓度抑制作用,SM-1的小肠吸收机制表现为被动扩散。
不同肠段对药物的吸收情况见Tab 4。SM-1在各肠段均有吸收,其在十二指肠的吸收优于其他肠段(P<0.05),而Ka和Peff在空肠、回肠与结肠间差异无显著性(P>0.05)。

Tab 3 K a and P eff of SM-1 in different concentrations(¯x±s,n=6)

Tab 4 K a and P eff of SM-1 in different intestine segments(¯x±s,n=4)
2.5 大鼠体内绝对生物利用度研究 大鼠静注及灌胃给予SM-1后的血药浓度-时间曲线如Fig 2。主要药动学参数见Tab 5。SM-1在大鼠体内的绝对生物利用度为29.3%。
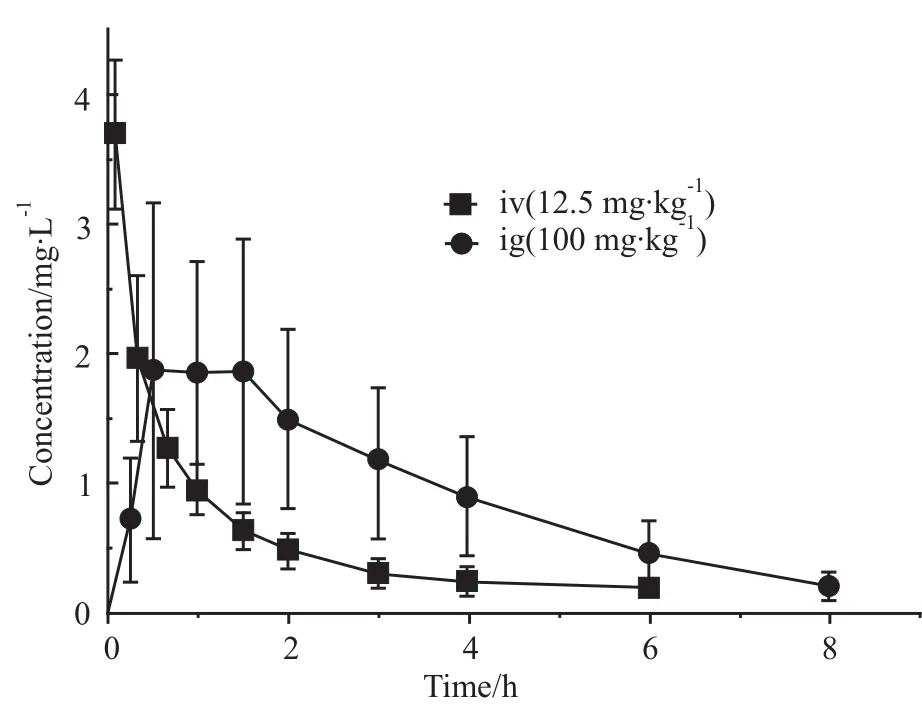
Fig 2 Plasma concentration-time profile of SM-1 in rats following ig 100 mg·kg-1 and iv 12.5 mg·kg-1(n=6)
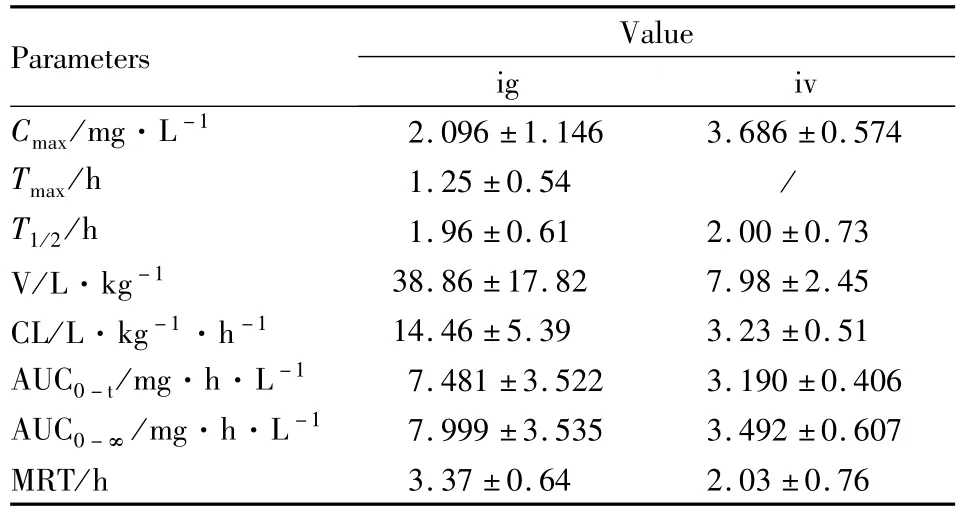
Tab 5 Main pharmacokinetic parameters of SM-1 in rats following ig 100 mg·kg-1 or iv 12.5 mg·kg-1(¯x±s,n=6)
3 讨论
3.1 Caco-2细胞双向转运 Caco-2细胞模型中[9],吸收良好的药物的 Papp>1×10-6cm·s-1;吸收较好的药物 Papp为0.1×10-6cm·s-1~1×10-6cm·s-1;而吸收差的药物(即吸收 <1%)的 Papp<1×10-7cm·s-1,据此可知 SM-1属于吸收良好的化合物。在Caco-2细胞模型对SM-1的双向转运实验中,不同浓度及转运方向间表观渗透系数均保持基本恒定,吸收以被动扩散为主。
3.2 在体肠吸收 文献报道[10],在体肠吸收实验中,当 Peff(rat)>5.9×10-5cm·s-1时,Fa(human)接近100%;当 Peff(rat)<3.32×10-5cm·s-1时,Fa(human)小于60%。本实验结果显示 SM-1的 Peff(rat)≈5.1×10-5cm·s-1,属于高渗透性药物。因此,根据体外及在体模型可以预示SM-1在体内吸收良好。在体肠吸收实验中,不同浓度SM-1的Ka与Papp差异无显著性,说明其在考察浓度范围内吸收不受自身浓度影响,主要以被动扩散机制进入体循环,且肠段平衡后每15 min Ka值基本不变,提示SM-1吸收符合一级动力学特征。在实验过程中,由于大鼠肠道会吸收一定的水分,导致灌流液体积发生改变,从而影响结果的准确性。研究者通常在灌流液中加入肠不吸收的物质(酚红,14C标记的 PEG)来校正体积[11],或者采用重量法来计算其体积。为了进一步降低实验的误差,本实验对灌流液密度进行了校正,校正后的密度为(1.025±0.045)kg·L-1,根据校正后的密度计算Ka和 Peff。
3.3 大鼠体内初步药动学 根据前期SM-1的药效学与毒理学研究结果,选择大鼠药动学研究的灌胃剂量为100μg·g-1。实验过程中考察了 CMCNa、HPMC、吐温-80对SM-1混悬液的助悬效果,结果发现含CMC-Na、HPMC的SM-1混悬液分散效果较差,而含0.5%吐温-80的SM-1混悬液均一性较好,沉降速度较慢,故选择其作为灌胃给药的助悬剂。由于SM-1在生理盐水中的溶解度约为4 g·L-1,在考察剂量下溶解不完全,需加入附加剂以增加其溶解度。实验发现SM-1在10%丙二醇中溶解度明显增加(丙二醇注射用溶剂范围为 10% ~60%),且大鼠在注射不含药的丙二醇-生理盐水时未出现不适现象。因此选择体积分数为0.1的丙二醇生理盐水作为注射用溶剂。然而,当 SM-1给药剂量为25μg·g-1时,大鼠出现血尿现象,进而降低给药剂量至12.5μg·g-1,此时血尿现象消失。因此,选择静脉注射给药剂量为12.5μg·g-1。
大鼠体内药动学研究显示SM-1口服吸收和消除较快,表观分布容积较大,血浆药物浓度和绝对生物利用度较低。结合前期体外研究结果,SM-1在酸性条件下极易降解,推测药物在胃液中的损失可能是导致生物利用度低的原因之一。因此,后期可考虑将其作为肠溶制剂开发。
4 结论
本研究为SM-1的成药性评价以及剂型设计提供了研究基础,但 SM-1在吸收转运过程中是否受到主动转运载体及细胞内吞作用等因素影响,口服后是否经过首过消除有待进一步研究。
参考文献:
[1] Peterson QP,Hsu D C,Goode D R,et al.Procaspase-3 activation as an anti-cancer strategy:structure-activity relationship of procaspaseactivating compound 1(PAC-1)and its cellular Co-Localization with caspase-3[J].J Med Chem,2009,52(18):5721-31.
[2] Karson SP,Grace W C,Jennifer M P,et al.Small-molecule activation of procaspase-3 to caspase-3 as a personalized anticancer strategy[J].Nat Chem Biol,2006,2(10):543-50.
[3] Peterson Q P,Goode D R,West D C,et al.PAC-1 activates procaspase-3 in vitro through relief of zinc-mediated inhibition[J].J Mol Biol,2009,388(1):144-58.
[4] Strand O A,Aziz G,Ali SF,et al.Synthesis and initial in vitro biological evaluation of two new zinc-chelating compounds:comparison with TPEN and PAC-1[J].Bioorg Med Chem,2013,21(17):5175-81.
[5] 关 溯.CaCo-2细胞模型-药物吸收研究的有效“工具”[J].中国药理学通报,2004,20(6):609-14.
[5] Guan S.CaCo-2 cell model——an effective tool for the research of drug absorption[J].Chin Pharmacol Bull,2004,20(6):609-14.
[6] 王来友,毕惠嫦,周洪波,等.花椒毒素在小肠中的吸收机制研究[J].中国药理学通报,2009,25(2):278-9.
[6] Wang L Y,Bi H C,Zhou H B,et al.Absorption mechanism of Xanthotoxin in small intestine[J].Clin Pharmacol Bull,2009,25(2):278-9.
[7] 宋 丽,张 宁,徐德生.芍药苷在CaCo-2细胞模型中吸收机制的研究[J].中草药,2008,39(1):41-4.
[7] Song L,Zhang N,Xu D S.Absorption mechanism of paeoniflorin across CaCo-2 monolayer mode[J].Chin Tradi Herbal Drugs,2008,39(1):41-4.
[8] 聂淑芳,潘卫三,杨星钢,等.对大鼠在体肠单向灌流技术中重量法的评价[J].中国新药杂志,2005,14(10):1176-9.
[8] Nie SF,Pan W S,Yang X G,et al.Evaluation of gravimetry in the rat single-pass intestinal perfusion technique[J].Chin New Drugs J,2005,14(10):1176-9.
[9] Artursson P,Palm K,Luthman K.Caco-2 monolayers in experimental and theoretical predictions of drug transport[J].Adv Drug Deliv Rev,1996,22(1-2):67-84.
[10]Milania P Z,Valizadeha H,Tajerzadehc H,et al.Predicting human intestinal permeability using single-pass intestinal perfusion in rat[J].Pharm Pharm Sci,2007,10(3):368-79.
[11]Dahan A,West B T,Amidon G L,et al.Segmental-dependent membrane permeability along the intestine following oral drug administration:Evaluation of a triple single-pass intestinal perfusion(TSPIP)approach in the rat[J].Eur J Pharm Sci,2009,36(2-3):320-9.
猜你喜欢
趣味(数学)(2021年9期)2022-01-19
载人航天(2021年5期)2021-11-20
天津医科大学学报(2021年3期)2021-07-21
中华养生保健(2020年7期)2020-11-16
中学生数理化(高中版.高二数学)(2020年2期)2020-04-21
中国当代医药(2018年12期)2018-06-16
中成药(2017年4期)2017-05-17
中国比较医学杂志(2017年3期)2017-01-17
中学生数理化·高三版(2016年9期)2016-05-14
中国比较医学杂志(2015年9期)2015-05-11
