
基于大鼠原位肠-肝血管灌流模型研究甘草酸的代谢
2014-05-14 11:21钟运鸣王素军黄丽花程漩格王桂香臧林泉
中国药理学通报 2014年4期
钟运鸣,王素军,曾 洁,黄丽花,程漩格,王桂香,臧林泉
甘草为豆科甘草属植物甘草(glycyrrhiza uralensis Fisch.)、胀果甘草(glycyrrhiza inflata Bat.)或光果甘草(glycyrrhiza glabra L.)的干燥根及根茎,有“国老”之美誉,是我国临床上常用的中药之一。甘草主要含三萜皂苷类和黄酮类。其中三萜皂苷类甘草酸(glycyrrhizin,GZ)含量最高,达6% ~14%,又名甘草甜素或甘草皂苷。近年来药理和临床研究发现甘草酸及其衍生物具有抗病毒、抗溃疡、抗变态反应、抗炎、抗肿瘤、保肝等多种生物活性[1],引起国内外学者的广泛关注。但有报道证明甘草酸的口服生物利用度极低(3.04%),且大部分被代谢成甘草次酸(glycyrrhetinic acid,GA)而产生高血压、电解质紊乱和假性醛固酮增多症等副作用[2-3]。然而,甘草酸在大鼠体内代谢时其肠肝各自发挥的作用,目前国内外尚未见报道。为此,本文首次尝试采用大鼠原位肠-肝血管灌流模型,观察甘草酸在大鼠肠道和肝脏中的处置,定量研究了甘草酸的吸收和代谢过程,并评价肠和肝这两个体内主要代谢器官在甘草酸代谢中的作用,从而为甘草酸的代谢靶点、药理作用、不良反应及临床合理用药提供参考依据。
1 材料与方法
1.1 仪器、试剂和动物 LC-20 AD高效液相色谱仪器(日本岛津);4000Q TRAP串联四极杆质谱仪(美国 Applied Biosystem公司);SUPERT21高速冷冻离心机(美国Sorvail公司);Milli-Q Gradient A10超纯水器(美国Millipore公司);501型超级恒温箱(上海市实验仪器厂);HL-2恒流泵(上海沪西仪器厂);甘草酸(批号:A0037),甘草次酸(批号:A0038),内标大黄素(批号:110756-200110)均购自中国药品生物制品检定所;色谱纯甲醇、色谱纯乙酸铵均购自美国Merck公司;其他试剂均为分析纯。SPF级S-D大鼠(200~250 g),♀♂各半,购于广州中医药大学实验动物中心,动物许可证号:scxk(粤)2008-0020。
1.2 实验方法
1.2.1 灌流液的配制 Krebs-Ringer NaHCO3缓冲液(K-R液):0.22 g NaH2PO4、7.8 g NaCl、0.37 g CaCl2、0.22 g MgCl2、0.35 g KCl、1.37 g NaHCO3、3.0 g葡萄糖加蒸馏水定容至1 000 ml,调节pH至7.4。内含3%右旋糖酐T-40、5% 牛血清白蛋白、10%洗过的大鼠红细胞、0.02%地塞米松和0.004%去甲肾上腺素。
1.2.2 样品处理和LC-MS/MS分析法 样品分析方法参照文献稍加改进[4]。样品处理:取样品100 μl于1.5 ml EP管中,精密加入浓度为40μg·L-1内标的甲醇-水溶液(V∶V=1∶1)300μl,涡旋振荡混匀,离心(14 000 r·min-1,20℃)30 min,取上清10μl进样分析。液相条件:色谱柱采用 Phenomenex Luna C18柱(2.00 mm×50 mm,5μm),流动相为10 mmol·L-1乙酸铵水溶液-甲醇(V∶V=95∶5),柱温25℃,体积流量为 0.2 ml·min-1。质谱检测条件:采用ESI源负离子模式、多反应检测(MRM)模式进行测定。检测离子对分别为 m/z 821.4→351.3(甘草酸)、m/z469.4→425.8(甘草次酸)和 m/z 269→225(大黄素),毛细管电压4 000 V,干燥气温度为330℃,喷雾压力55 pis,驻留时间0.1 s。
1.3 模型的建立
1.3.1 大鼠原位单向肠-肝血管灌流模型(in situ single pass vascularly perfused rat intestine-liver model,IPIL) 参照我们课题组以前的实验方法[5-6],实验前先用空白灌流液平衡系统15 min后,将含有甘草酸(5 mg·L-1)的灌流液恒定流速(10 ml·min-1)从上肠系膜动脉输入,经过肝静脉流出,不形成循环。分别从门静脉和肝静脉每隔5 min吸取250μl灌流液置于1.5 ml EP管,持续1 h。胆汁每10 min为一个取样点,同样持续1 h。样品处理后立即进样分析。实验手术图解如Fig 1。
1.3.2 大鼠原位循环肠血管灌流模型(in situ circulated vascularly perfused rat intestine model,IPI)参阅我们课题组以前的模型[5],灌流液从上肠系膜动脉流入,从门静脉流出,使得肠血管与导管、储液池构成一闭合通路,固定插管后将体积流量提高至7.5 ml·min-1,整个实验过程中用白炽灯加热控制温度。实验前先用空白灌流液平衡15 min并以此作为实验零点,于十二指肠给予2 mg甘草酸,分别于0、5、15、30、45、60、75、90、105、120min时从储液池吸取250μl灌流液置于1.5 ml EP管中,在小肠入口和出口处引流收集肠腔液。每次取样后立即补充同体积的空白灌流液(灌流总体积为100 ml)。样品处理后立即进样分析。
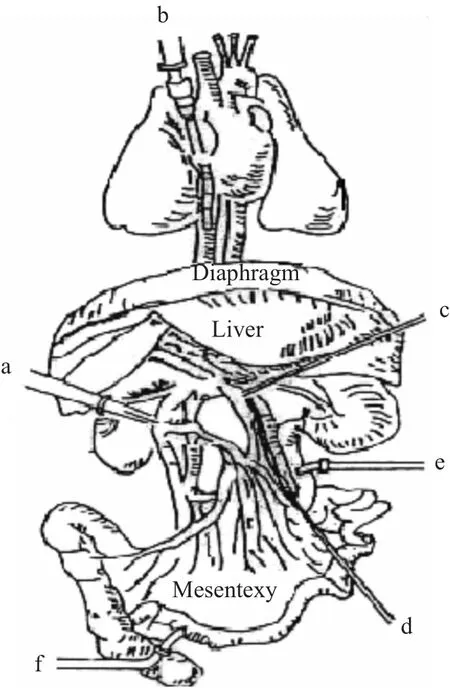
Fig 1 Surgical cannulation for in situ vascularly perfused rat intestine-liver model
1.4 数据处理 数据用¯x±s表示,肠稳态提取率(steady state extraction ratio of the intestine,Ei)和肝稳态提取率(steady state extraction ratio of the liver,Eh)分别用公式(1)和(2)计算;肠清除率(clearance ratio of the intestine,CLi)、肝清除率(clearance ratio of the liver,CLh)和肠肝总清除率(total clearances,CLt)分别用公式(3)、(4)和(5)计算;IPI中以灌流池中目标化合物药量的对数对取样时间进行线性回归,得回归方程,根据回归方程的斜率求算吸收速率Ka。
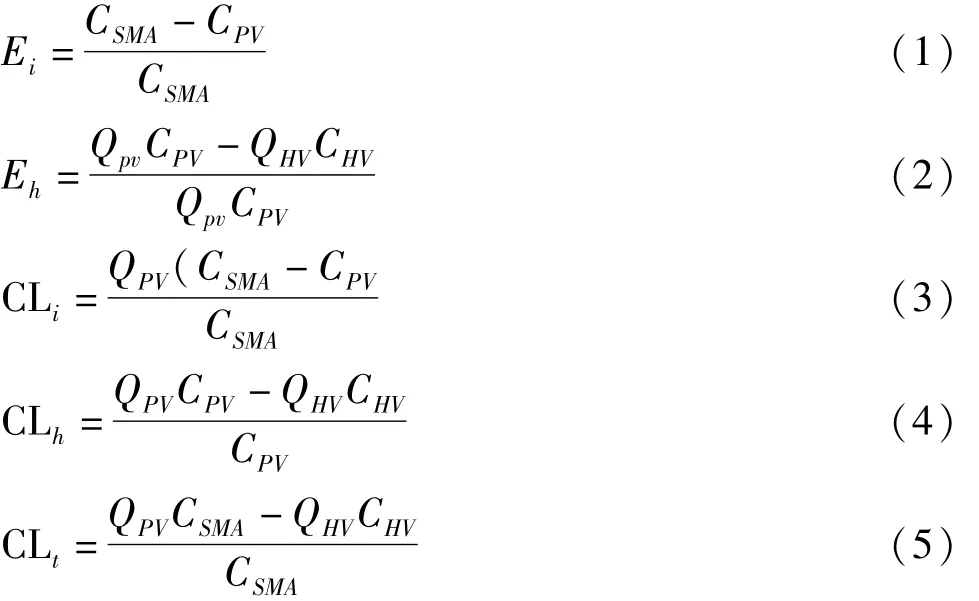
其中CSMA、CHV和CPV分别表示上肠系膜动脉、肝静脉、门静脉的稳态药物浓度,QPV和QHV分别表示门静脉和肝静脉的药物灌流流速。
2 结果
2.1 甘草酸单次通过肠-肝血管灌流模型的动力学过程 单次通过大鼠肠-肝血管灌流后,甘草酸形成的稳态动力学过程如Fig 2。5 mg·L-1的甘草酸以10 ml·min-1流速通过上肠系膜动脉进行灌流。当流出产物形成稳态后(30 min后),甘草酸在灌流液中的变化速率为零。经过肠肝作用后,在肝静脉流出液和门静脉流出液中均只检测到甘草酸而没有检测到甘草次酸。在胆管插管收集的胆汁中同样只检测到甘草酸,且甘草酸在胆汁中所占的比例较高(Fig 2),在60 min时甘草酸在胆汁中所占比例已达到85%(Fig 3)。灌流液和胆汁中流出的甘草酸均在30 min左右达到稳态。
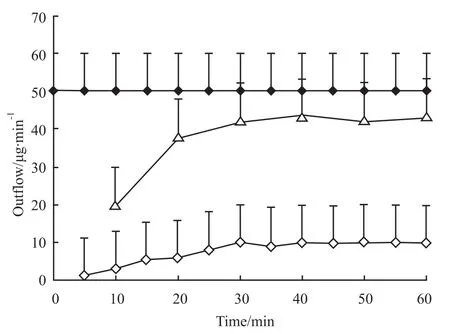
Fig 2 Disposition of GZ during its single passage through the perfusate rat intestine-liver model(¯x±s,n=6)
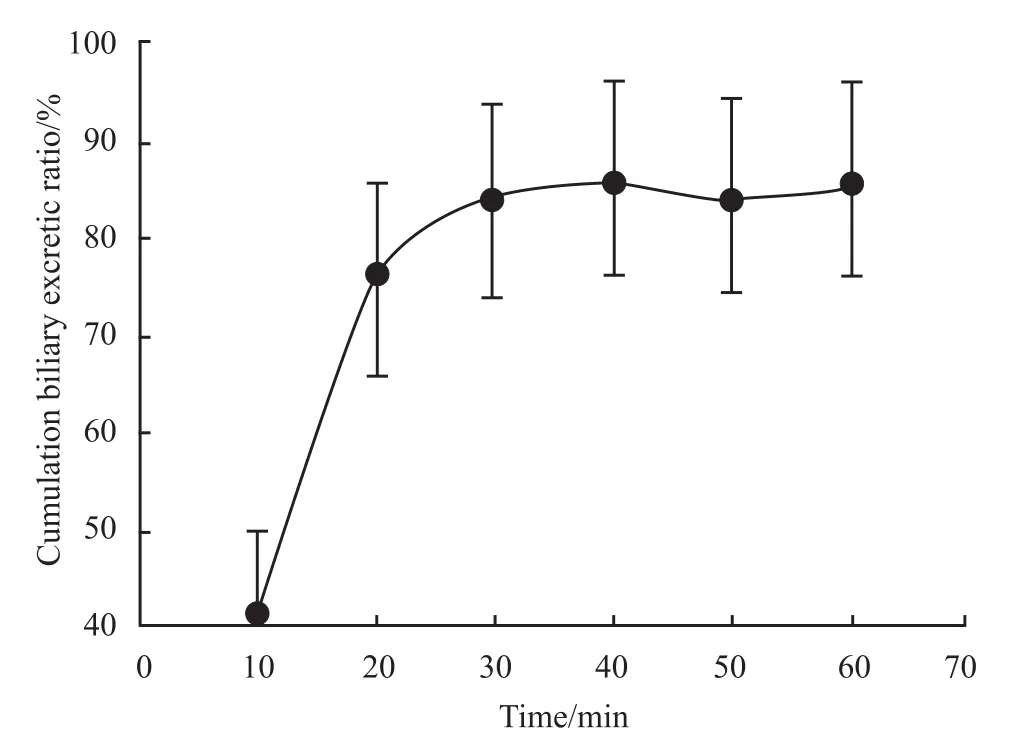
Fig 3 Cumulative biliary excretion ratio of unchanged GZ excreted in bile(¯x±s,n=6)
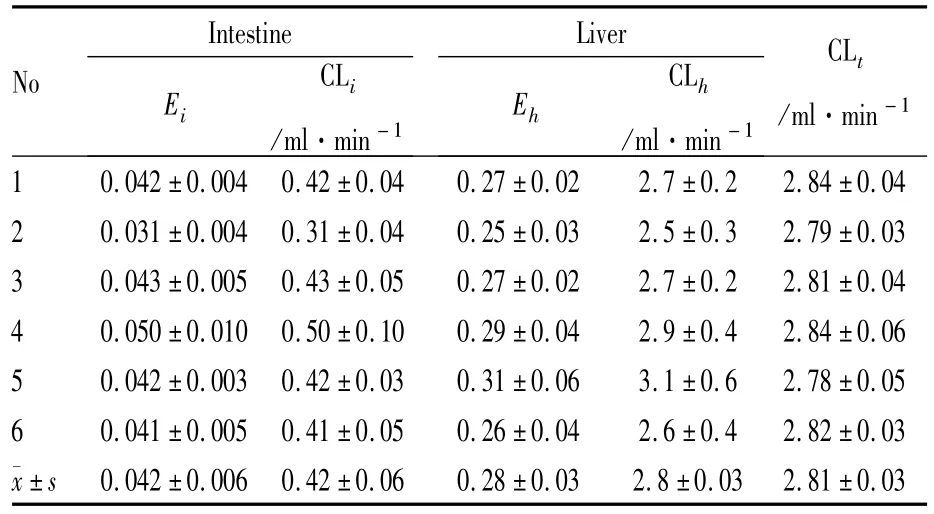
Tab 1 Metabolic parameters of GZ in the once-through vascularly perfused rat intestine-liver preparation(¯x±s,n=6)
2.2 甘草酸大鼠十二指肠给药后肠血管灌流模型循环灌流的处置过程 样品处理后经LC-MS/MS测定如Fig 4。上述色谱系统中,无内源性物质干扰灌流液,检测物色谱行为较佳,甘草酸、甘草次酸和大黄素保留时间分别约为2.74、3.56和3.12 min。甘草酸大鼠十二指肠给药后及其代谢物甘草次酸在灌流液中的动力学曲线如Fig 5。2 mg甘草酸十二指肠给药后,0~80 min时甘草酸吸收较快,80 min后开始趋于平稳;120 min时的浓度为(0.42±0.06)mg·L-1,经计算得出甘草酸吸收量为2.01%(120 min时灌流液中甘草酸含量与肠道给药量的比值)。代谢产物甘草次酸在灌流5 min后能检测到,甘草次酸20~80 min上升较快,120 min时浓度为(8.45±0.76)mg·L-1。
由于甘草酸在循环灌流系统中仅仅通过代谢而消除,甘草酸和其代谢产物均未离开系统。储液池中甘草酸及其产物的浓度-时间动力学曲线具有静注单室模型和血管外途径单室模型的动力学特征,故我们选用药动学计算软件(3P97)对甘草酸和产物甘草次酸的消长动力学过程进行拟合,曲线的实测值与拟合值间有良好的相关性(R>0.9),由此得到的主要药动学参数(Tab 2)。
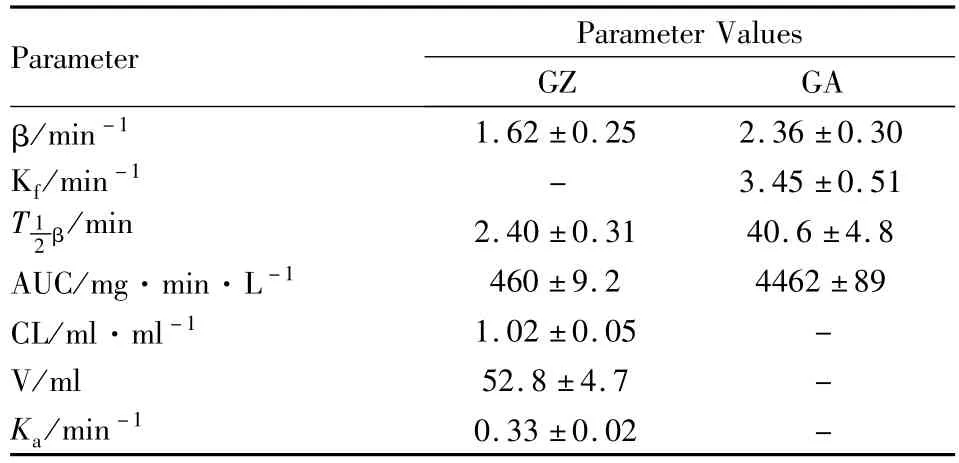
Tab 2 Kinetic parameters of GZ and its metabolite(GA)in the intestinal perfusion(¯x±s,n=6)
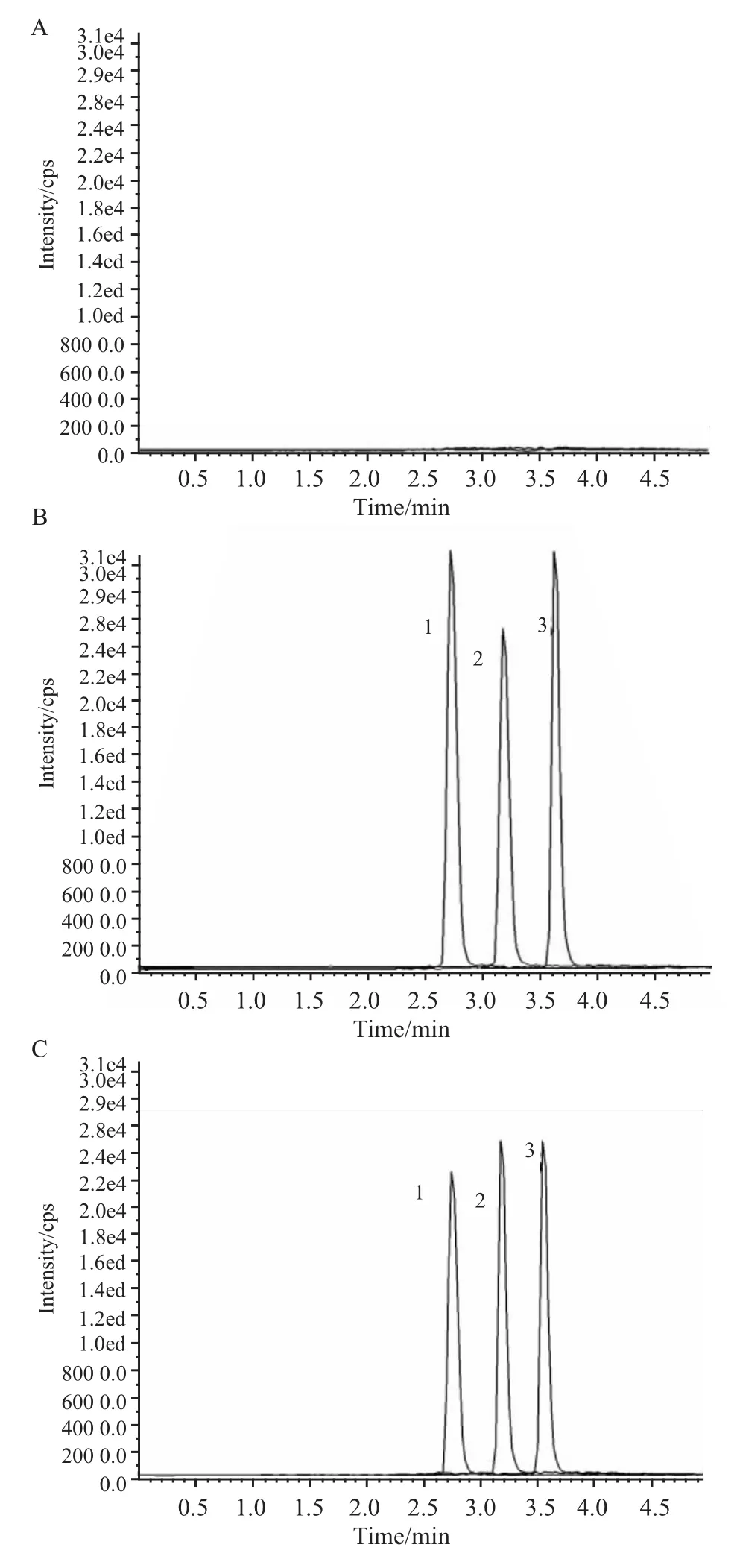
Fig 4 LC-MS/MSchromatograms of perfusion samples
3 讨论
大鼠原位肠-肝血管灌流模型具有诸多优点,如保持了实验中肠肝的完整性,同时避免了药物的广泛分布、可分析各种外加因素对药物代谢的影响、将肠灌流和肝灌流结合一起,不仅可以评价肠肝的共同作用亦可评价各自作用亦可研究肠道菌群和酶对药物代谢的影响。因而这一模型在药物的首过效应、吸收和代谢等研究中具有广阔的应用前景。
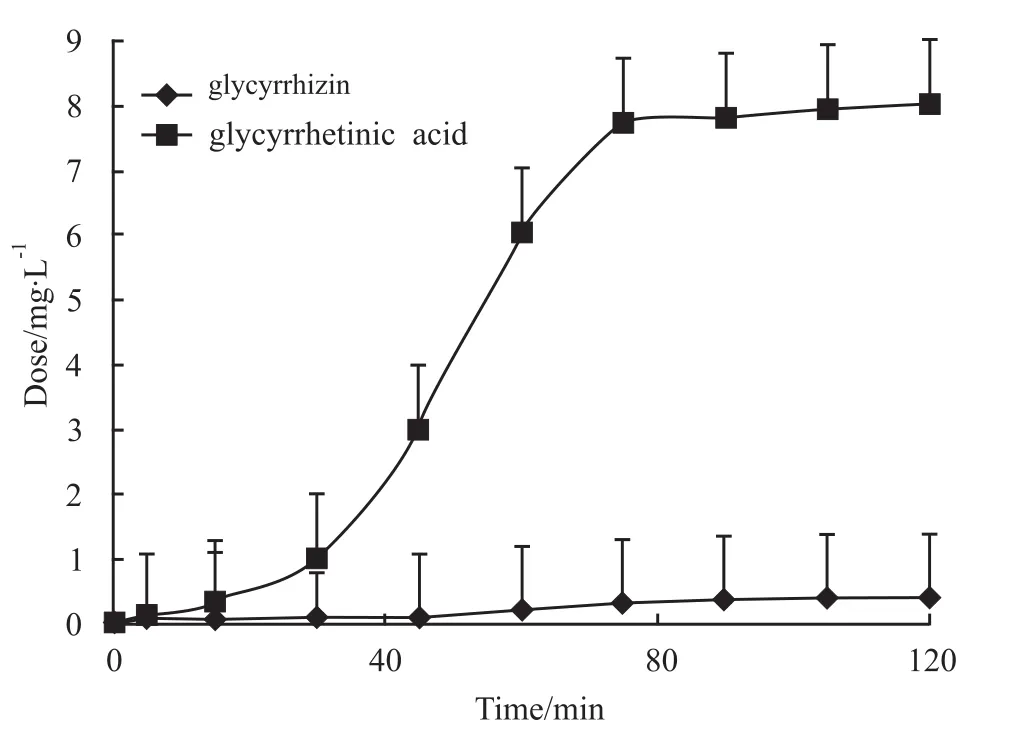
Fig 5 Disposition of GZ during its recirculating intestinal perfusion after intraduodenal administration(¯x±s,n=6)
本文在前期工作[5-6]的基础上利用该模型研究甘草酸在大鼠体内的代谢,并评估了肠肝在甘草酸代谢中的作用。在单向肠-肝血管灌流实验中,甘草酸的稳态肝提取率(28.0±3.0)%,是稳态肠提取率(4.2±0.6)%的7倍,且在肝静脉流出液和门静脉流出液均只检测到甘草酸的存在,这些表明上肠系膜动脉给予甘草酸后,甘草酸未在肝脏中被代谢。由于甘草酸在肝脏中不是被代谢成甘草次酸,所以甘草酸的肝首过消除不是造成其生物利用度低的主要原因,而是因为肠道菌群对其的大量代谢。同时收集的胆汁中我们检测到大量的甘草酸,1 h后甘草酸在胆汁中所占比例已达到85%(Fig 3),说明从门静脉进入的甘草酸很大一部分被分泌到胆汁中。从而佐证了甘草酸从门静脉注射给药后很难在系统静脉血中检测到甘草次酸[7-8]。在循环肠血管灌流中,十二指肠给予2 mg甘草酸后,通过检测储液池中的灌流液中发现甘草酸与甘草次酸同时存在(Fig 4),计算得出甘草酸的一级吸收速率常数和消除半衰期β分别为(0.33±0.06)min-1和(2.40±0.31)min。有研究表明[9]甘草次酸的一级吸收速率常数在(1.53~1.87)min-1间且甘草次酸的表观渗透系数是甘草酸的10倍以上。上述肠血管灌流实验中甘草酸的吸收速率常数小于甘草次酸吸收速率常数参考值,而且灌流120 min后,甘草酸的吸收量仅为2.01%,表明甘草酸在肠道吸收差代谢快,大部分转变为甘草次酸。通过检测肠腔液体,发现存在大量甘草次酸,进一步说明甘草酸在肠道菌群或肠道酶存在下被大量代谢为甘草次酸,这结果与文献相符合[10-11]。
综上所述,原位肠-肝血管灌流模型揭示了口服甘草酸后,甘草酸很难被吸收而且主要在肠道中被菌群或酶代谢。同时原位肠-肝灌血管流模型在研究药动学方面具有很大的潜力和广阔的应用前景。不仅可以评价机体对药物的首过效应,还可以了解肠道、肝脏在这一过程中的作用及各自的贡献度。
参考文献:
[1] Ming L J,Yin A C.Therapeutic effects of glycyrrhizin acid[J].Nat Prod Commun,2013,8(3):415-8.
[2] Isbrucker R A,Burdock G A.Risk and safety assessment on the consumption of Licorice root(glycyrrhiza sp),its extract and powder as a food ingerdient,with emphasis on the pharmacolocy and toxicology of glycyrrhizin[J].Requl Toxicol Pharmacol,2006,46(3):167-92.
[3] 杨锦南,朱 明.甘草次酸及其衍生物药理作用研究进展[J].中国药理学通报,1997,13(2):110-4.
[3] Yang J N,Zhu M.Progresses in pharmacological effects of glycyrrhetinic and its derivatives[J].Chin Pharmacol Bull,1997,13(2):110-4.
[4] Wang Y,Xu C,Wang P,et al.Pharmacokinetic comparison of different combination of Shaoyao-Gancao-Decoction in rat:simultanecous determination of ten active constituents by HPLC-MS/MS[J].J Chromatogr B Analyt Technol Biomed Life Sci,2013,1(932):76-87.
[5] 王素军,阮金秀,张振清,等.大鼠原位肠-肝灌流模型研究小补心汤总黄酮体内效应物质[J].中国药理学通报,2011,27(6):885-6.
[5] Wang S J,Ruan J X,Zhang Z Q,In vivo bioactive substance study of Xiao Bu Xin Tang tptal flavoniods extracts by in suit perfused rat intestine-liver moldel[J].Chin Pharmacol Bull,2011,27(6):885-6.
[6] 莫李立,王素军,杨本坤.阿魏酸在Caco-2细胞模型的通透性及其在大鼠体内吸收特征性研究[J].中草药,2012,5(43):947-51.
[6] Mo L L,Wang SJ,Yang B K.Permeability of ferulic acid in Caco-2 cell model and its absorption properties in rats in vivo[J].Chin Tradit Herb Drugs,2012,5(43):947-51.
[7] Ploeger B,Mensinga T,Sips A,et al.The pharmacokinetics of glycyrrhizic acid evaluated by physiologically based pharmacokinetic modeling[J].Drug Metab Rev,2001,33(2):125-47.
[8] 黎国富,杨 劲,华小懿,等.Ussing Chamber模型研究甘草酸二铵经大鼠肠粘膜的转运和代谢[J].中国中药杂志,2010,35(17):2261-5.
[8] Li G F,Yang J,Hua X Y,et al.Tranmembrance transport and metablosim of diammonium glycyrrhizinateacross rat small intestine in Ussing Chamber[J].Chin J Chin Mat Med,2010,35(17):2261-5.
[9] Ishida S,Sakiya Y,Taira Z.Disposition of glycyrrhizin in the perfused Intestine of rats[J].Biol Pharm Bull,1994,17(12):960-9.
[10]Ishida S,Sakiya Y,Ichikawa T,Awazu S.Pharmacokinetics of glycyrrhetic acid,a major metabolite of glycyrrhizin,in rats[J].Chem Pharm Bull,1989,37(9):210-7.
[11]宋 丽,徐璐扬,张 宁.大鼠肠内微生物对甘草酸代谢的影响[J].上海中医药杂志,2008,42(9):70-2.
[11]Song L,Xu L Y,Zhang N.Effects of intestine bacteria on metabolish of glycyrrizin in rat[J].Shanghai J Tradit Chin Med,2008,42(9):70-2.
猜你喜欢
实用手外科杂志(2022年2期)2022-08-31
陶瓷学报(2021年5期)2021-11-22
载人航天(2021年5期)2021-11-20
天津医科大学学报(2021年3期)2021-07-21
中华养生保健(2020年7期)2020-11-16
天然产物研究与开发(2018年5期)2018-06-13
中成药(2018年5期)2018-06-06
中国中西医结合皮肤性病学杂志(2016年4期)2016-07-18
中国当代医药(2015年33期)2015-03-01
中国当代医药(2015年10期)2015-03-01
