
高效液相色谱法测定紫杉醇亚微乳含量
2014-05-02 01:28夏学军王艳宝徐佳茗张鹏霄金笃嘉刘玉玲
中国药业 2014年3期
夏学军,王艳宝,徐佳茗,张鹏霄,金笃嘉,刘玉玲
(中国医学科学院北京协和医学院药物研究所·药物传输技术及新型制剂北京市重点实验室,北京 100050)
紫杉醇亚微乳是我所研制的不含Cremophor EL、载药量高、可耐热压灭菌、长期贮存稳定的具有自主知识产权的水包油型亚微乳输液[1]。动物试验表明,与市售紫杉醇注射液相比,亚微乳可显著降低过敏反应和毒性。在耐受剂量下,紫杉醇亚微乳比市售注射液对人乳腺癌MDA-MB-231裸鼠异种移植性肿瘤治疗效果更好。目前,该药已完成临床前研究,正准备申报临床。为了控制该药的质量,笔者参考2010年版《中国药典(二部)》紫杉醇标准[2],建立了紫杉醇亚微乳含量测定的高效液相色谱(HPLC)法,现报道如下。
1 仪器与试药
Agilent 1200型高效液相色谱仪,包括G1322A型真空脱气机、G1310A型四元泵、G1329A型自动进样器、G1314B VWD型检测器和 Chemstation型色谱工作站(Agilent公司)。紫杉醇对照品(中国药品生物制品检定所,批号为100382-200301);三杉尖宁碱(紫杉醇杂质Ⅰ,中国药品生物制品检定所,批号为100926-200701);7-表-10-去乙酰基紫杉醇(紫杉醇杂质Ⅱ,中国药品生物制品检定所,批号为100925-200701);紫杉醇亚微乳(批号为 20110409-1,20110409-2,20110415,规格为 1.176 g/L);空白亚微乳(辅料,按紫杉醇亚微乳的处方和工艺不加主药制备而得,批号为 20100716);无水乙醇(色谱纯,J.T.Baker公司);乙腈(色谱纯,J&K Chemical Ltd.);甲醇(色谱纯,Fisher Scientific 公司);其他试剂均为分析纯。
2 方法与结果
2.1 色谱条件与系统适用性试验
色谱柱:Zorbax XDB C18柱(250mm ×4.6mm,5 μm);流动相:甲醇-水-乙腈(23 ∶41 ∶36);流速:1.5 mL/min;进样量:10 μL;柱温:40℃;检测波长:227 nm。分别取紫杉醇、三杉尖宁碱、7-表-10-去乙酰基紫杉醇对照品适量,用无水乙醇溶解制成一定浓度的溶液,分别取10 μL注入高效液相色谱仪,进行色谱峰定位。另取紫杉醇、三杉尖宁碱、7-表-10-去乙酰基紫杉醇对照品适量,用无水乙醇溶解制成质量浓度分别为 0.5,2.5,2.5 μg/mL 的混合溶液,作为系统适用性溶液,取 10 μL 注入高效液相色谱仪测定。结果三杉尖宁碱及7-表-10-去乙酰基紫杉醇与紫杉醇的相对保留时间分别为 0.89,1.09 min,分离度分别为 2.40,1.67,色谱图见图 1。
2.2 溶液制备
取紫杉醇适量,精密称定,加无水乙醇溶解制成质量浓度为1.2 g/L 的贮备溶液。分别精密量取 0.8,1.0,1.2 mL(相当于供试品溶液测定浓度的80%,100%,120%)各3份,置25 mL容量瓶中,分别加入空白亚微乳(辅料)0.8,1.0,1.2 mL,加无水乙醇稀释至刻度,摇匀,制成相当于紫杉醇含量80%,100%,120%的溶液,即得空白对照溶液。
2.3 方法学考察
专属性试验:取空白亚微乳(辅料)1 mL,置25 mL容量瓶中,用无水乙醇稀释至刻度,摇匀,作为辅料溶液,取10 μL注入高效液相色谱仪。结果表明,辅料对亚微乳的含量测定无干扰。色谱图见图 1。
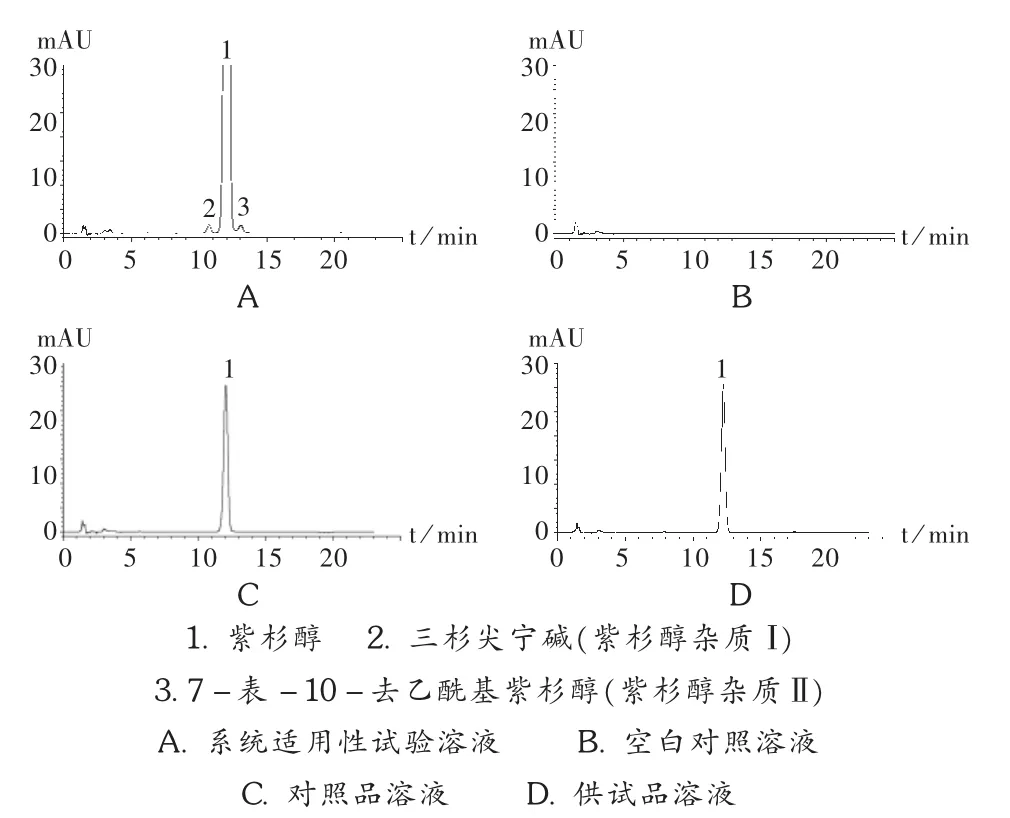
图1 高效液相色谱图
线性关系考察:取紫杉醇适量,精密称定,加无水乙醇溶解制成质量浓度为 1.2 g/L 的溶液。分别精密量取 0.2,0.5,1,1.5,2 mL,置25 mL容量瓶中,加无水乙醇稀释至刻度,摇匀,制得质量浓度分别为 9.6,24,48,72,96 μg/mL 的标准溶液。分别取 10 μL 注入高效液相色谱仪,连续进样3次。以质量浓度(X,μg/mL)为横坐标、平均峰面积(Y)为纵坐标进行线性回归,得回归方程 Y=13.788 7 X-3.424 4, r=0.999 9(n=5)。结果的 RSD 分别为0.12% ,0.12% ,0.10% ,0.16% ,0.08% ,表明紫杉醇样品质量浓度在9.6~96 μg/mL范围内与峰面积呈良好线性关系。
定量限检测:紫杉醇质量浓度为 0.3 μg/mL,进样量为 10 μL,其峰信号约为噪音的10倍,计算得该方法的定量限为3 ng。
重复性试验:取同一紫杉醇亚微乳样品6份,按拟订方法测定紫杉醇含量。结果,样品含量测定的 RSD为0.31%(n=6),表明方法重现性良好。
稳定性试验:取供试品溶液 10 μL,分别于 0,2,4,6,8,10,12 h时进样测定,记录峰面积。结果的 RSD为0.16%,表明供试品溶液在12 h内稳定。取紫杉醇适量,精密称定,加无水乙醇溶解制成质量浓度为 1.2 g/L 的贮备溶液。分别精密量取 0.8,1.0,1.2 mL(相当于供试品溶液测定浓度的80%,100%,120%)各3份,置25 mL 容量瓶中,分别加入空白亚微乳(辅料)0.8,1.0,1.2 mL,加无水乙醇稀释至刻度,摇匀,制成相当于紫杉醇含量80%,100%,120%的溶液,即得空白对照溶液。按拟订方法测定,计算回收率。结果见表1。
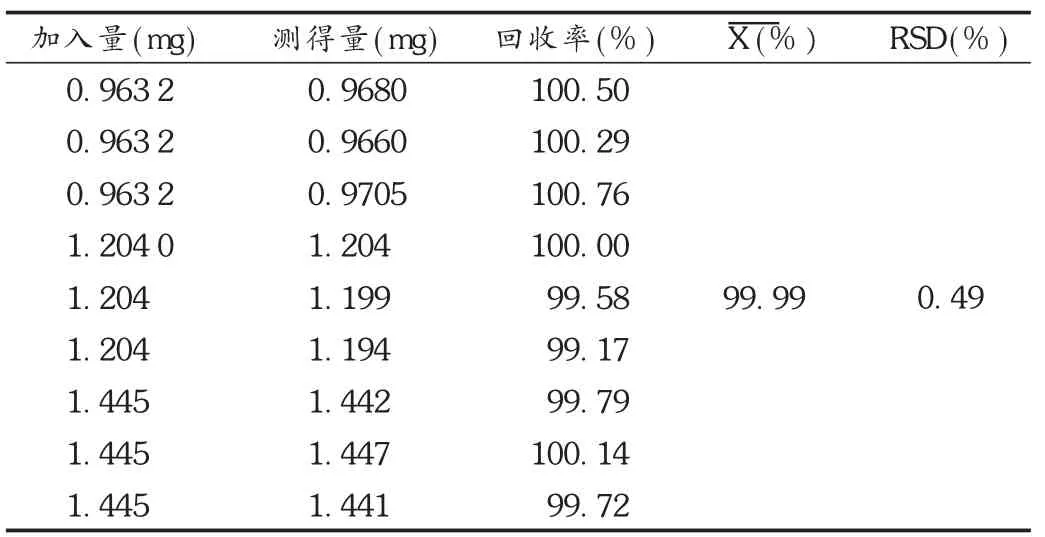
表1 回收率测定结果(n=9)
2.4 样品含量测定
精密量取本品1 mL,置25 mL容量瓶中,用无水乙醇稀释至刻度,摇匀,作为供试品溶液;另取紫杉醇对照品约22mg,精密称定,置10 mL容量瓶中,用无水乙醇稀释至刻度,振摇使溶解,精密量取1 mL,置50 mL容量瓶中,用无水乙醇稀释至刻度,摇匀,作为对照品溶液。按拟订色谱条件,分别取10 μL注入高效液相色谱仪,记录峰面积,采用外标法计算含量。结果,批号为20110409-1,20110409-2,20110415的样品中,紫杉醇的含量分别为标示量的 95.2% ,96.1% ,97.8% 。
3 讨论
2010年版《中国药典(二部)》收载了紫杉醇药品标准,其含量测定项下的系统适用性试验中,规定了紫杉醇峰与三杉尖宁碱(杂质Ⅰ)峰及7-表-10-去乙酰基紫杉醇(杂质Ⅱ)的分离度均应大于1.0。在流动相筛选时,在现有色谱条件下,对目前紫杉醇含量测定通常采用的3种流动相系统(甲醇-水、乙腈-水、甲醇-乙腈-水)进行了比较。结果发现,以甲醇-水、乙腈-水系统为流动相时,调整流动相比例使紫杉醇的保留时间在12 min范围内,方法的系统适用性均不能满足中国药典要求。
在紫杉醇亚微乳研究中,我们采用建立的HPLC有关物质检查法进行紫杉醇亚微乳及空白亚微乳(辅料)的强制降解试验、影响因素试验、加速试验及长期试验时发现,辅料对紫杉醇测定无干扰,亚微乳降解后主要产生2个杂质,经与美国药典收载的紫杉醇标准中各个杂质峰的相对保留时间进行比较,并与杂质对照品的保留时间进行比对,鉴定降解产物为10-去乙酰基紫杉醇及7-表-紫杉醇。故在拟订色谱条件下,试验了10-去乙酰基紫杉醇及7-表-紫杉醇对紫杉醇含量测定的干扰,结果10-去乙酰基紫杉醇及7-表-紫杉醇与紫杉醇的相对保留时间分别为 0.64,1.71 min,降解产物不干扰主成分测定。因此,按照 2010年版《中国药典(二部)》紫杉醇标准含量测定项下规定的系统适用性试验,即可保证本品测定方法的专属性符合要求。
亚微乳是由油、水、表面活性剂组成。由于药物与油脂分离困难,目前亚微乳含量测定供试品溶液制备时,除少量文献采用以高温、碱、盐等破乳后再用有机溶剂提取药物的方法外,大多采用直接用有机溶剂溶解稀释的方法。由于紫杉醇对高温、碱不稳定,我们前期曾参考文献[3],对盐破乳后再用有机溶剂提取的方法进行了研究,但未能同时获得紫杉醇与油脂的完全分离及合格的回收率。因此,本方法采用有机溶剂溶解稀释的方式制备供试品溶液。对有机溶剂进行的筛选表明,在测定浓度下,甲醇、乙腈均不能溶解紫杉醇亚微乳,无法满足紫杉醇测定浓度的要求,故选择无水乙醇作为本品的测定溶剂。
参考文献:
[1] Xia XJ,Guo RF,Liu YL,et al.Formulation,characterization and hypersensitivity evaluation of anintravenous emulsion loaded with a paclitaxel-cholesterol complex[J].Chem Pharm Bull(Tokyo),2011,59(3):321-326.
[2] 国家药典委员会.中华人民共和国药典(二部)[M].北京:中国医药科技出版社,2010:1 007-1 008.
[3] 李 萌,王杏林,葛志强,等.HPLC法测定依托泊苷亚微乳中的药物含量[J].中国新药杂志,2007,16(17):1 390-1 392.
猜你喜欢
陕西师范大学学报(自然科学版)(2022年5期)2022-11-09
发明与创新(2022年31期)2022-11-03
中国医药科学(2022年5期)2022-05-05
中国化妆品(2022年3期)2022-03-26
科海故事博览·中旬刊(2020年3期)2020-03-15
纺织服装流行趋势展望(2020年1期)2020-02-01
安徽化工(2018年5期)2018-10-23
纺织服装流行趋势展望(2016年6期)2016-05-04
纺织服装流行趋势展望(2016年4期)2016-05-04
纺织服装流行趋势展望(2016年1期)2016-05-04
