
Genetic diversity and gene structure of mitochondrial region of Anopheles minimus (Diptera: Culicidae) - major malaria vector of North east India
2014-04-20 08:23PrafullaDuttaSirajAhmedKhanRashmeeTopnoPritomChowdhuryMayuriBaishyaPurvitaChowdhuryJagadishMahanta
Prafulla Dutta, Siraj Ahmed Khan, Rashmee Topno, Pritom Chowdhury, Mayuri Baishya, Purvita Chowdhury, Jagadish Mahanta
Regional Medical Research Centre, ICMR (NE Region), Dibrugarh, Assam, India
Genetic diversity and gene structure of mitochondrial region of Anopheles minimus (Diptera: Culicidae) - major malaria vector of North east India
Prafulla Dutta*, Siraj Ahmed Khan, Rashmee Topno, Pritom Chowdhury, Mayuri Baishya, Purvita Chowdhury, Jagadish Mahanta
Regional Medical Research Centre, ICMR (NE Region), Dibrugarh, Assam, India
Objective:To depict mitochondrial genetic variation for the first time among Anopheles minimus (An.minimus) (Diptera: Culicidae) species from two malaria endemic states of NE India.Methods:Phylogeographic analysis was carried at 9 out of 12 sites of An.minimus confirmed malaria endemic places.Results:All sequences were Adenine-Thymine rich regions. Transitions were observed in 6 sequences where 5 mutations were synonymous substitutions and in 1 case non synonymous mutation was observed. Three distinct clusters of haplotypes were generated. Haplotype diversity and low nucleotide diversity were studied. Overall negative values obtained from Tajima’s D test and Fu'sFStest indicate a recent genetic population expansion. Network analysis has explained sequence diversity that was also shown by mutations in 6 sequences.
Conclusions:High genetic diversity observed within the populations of An.minimus species has several possible implications for vector control in the region.
ARTICLE INFO
Article history:
Received 24 September 2014
Received in revised form 10 October 2014
Accepted 15 November 2014
Available online 20 December 2014
Anopheles minimus
Cytochrome oxidase Ⅱ
Haplotype
Mutation
Substitution
1. Introduction
Malaria, an arthropod borne disease is endemic in many parts of the world including parts of America, Asia and Africa. According to a WHO report (2005), almost 300 million cases of malaria occur worldwide and more than a million people die due to malaria every year[1]. It is a common problem in India (latitude- 8°4’ N to 37°6’ N; longitude-68°7’ E to 97°25’ E), which contributes about 70% of malaria occurring in the South East Asian Region[2]. The National Vector Borne Disease Control Program (NVBDCP) of India reported -1.6 million cases and -1 100 malaria deaths in 2009. However, actual numbers are grossly underestimated and number of malaria cases per year may be between 9 to 50 times higher[3]. Malaria is endemic in North east region (NER) of India (latitude- 21°58’ N to 29°30’N and longitude- 88°3’ E to 97°30’ E). NER, (population 28.5 million) comprising of forests, forest fringes and extensive hill ranges and peninsulas provide a conducive environment for malaria transmission[4]. The NER comprises of eight states of which the highly malaria endemic states are Assam, Arunachal Pradesh (AP), Meghalaya, Mizoram and Tripura. The state of Assam and AP alone contributes 42% and 12% of malaria cases respectively in the NER[5]. Assam alone shares more than 5% of cases reported in India annually[6]. The present study was undertaken in two malaria endemic states -Assam (latitude- 24° 8’N to 28°2’ N; longitude- 89°42’E to 96°E) and AP (latitude- 26°30’ N to 29°30’ N; longitude- 91°30’ E to 97°30’ E). Human Malaria is caused by five species of parasitic protozoans within the genusPlasmodiumtransmitted by Anopheline mosquitoes. In NER, there are reportedly two main vectors-Anopheles dirus(the monsoon species) andAnopheles minimus (An. minimus) (the perennial species).An.minimushas been incriminated as vector from these two states[7,8].An.minimussensu lato (Myzomyia series, Funestus group) comprises of three sibling species-An.minimus(formerlyAn. minimusA),Anopheles harrisoni(formerlyAn.minimusC) andAnopheles yaeyamaensis(formerlyAn. minimusE). From ourstudy areas, out of the sibling complex, onlyAn.minimus sensu stricto (s.s.) species has been incriminated as vector species from the two states. However gene population analysis ofAn.minimus s.s.from the region has not been studied yet. Molecular markers -ribosomal DNA (ITS2 and D3) and mitochondrial markers (COⅠ, COⅡ, ND4 and ND5) were employed for genetic characterization of the vector species. The mitochondrial DNA (mtDNA) markers are often used for phylogenetic and population genetics analyses due to high information content at different evolutionary levels, including variation within and between populations. Coding content conservation, maternal inheritance, high mutation rate, presence of variable regions adjacent to highly conserved sites suitable for primer design and ease of amplification associated with a high copy number within the cell make mtDNA the molecule of choice for evolutionary studies. It has been widely utilized in the analyses of patterns of molecular evolution and as a marker for phylogeographic and phylogenetic inference. In mosquitoes, mtDNA sequences have been used to resolve relationships at various levels of divergence and to estimate dates of branching of major lineages. MtDNA have also been used for PCR-based identification of species belonging to cryptic species complexes[9]. The present study aimed at understanding the phylogeographic variation ofAn.minimusspecies within the study area.
2. Materials and methods
2.1. Mosquito sampling
Whole- night mosquito collections in human dwellings were carried out during study period from 17 sites in Assam and 8 sites in AP during 2008-2011 using dry cell operated Centers for Disease Control (CDC) miniature light traps. Collected mosquitoes were identified using standard Anopheline keys[10]. Mosquitoes were then stored individually in capped plastic vials containing silica gel and stored at 4 ℃ until further processing.
2.2. DNA extraction and diagnostic ASPCR assay
The identified mosquito was dissected into 3 parts, viz; head + thorax, abdomen and wings. The head + thorax and abdomen sections were homogenized separately by using tissue homogenizer (Axygen). Genomic DNA was extracted from first two parts by using FTA Classic card nucleic acid extraction technology (Whatman) for all samples by method of Mohantyet al[11].
Confirmation of morphologically identified Anophelines was done by Allele Specific PCR (ASPCR) after DNA extraction. ASPCR was done by following the method of Phucet al[12].
2.3. Gene amplification and sequencing of COⅡregion
ASPCR identifiedAn. minimusspecies were randomly chosen from all collection sites for molecular identification and phylogenetic informations using mitochondrial marker-COⅡ. Mitochondrial COⅡ gene of samples was amplified as per protocol of Morganet al[13]. Reaction volume was of 25 μ L containing 1×volume of Promega PCR Master Mix (2×) and 0.24 μM of each primers. Thermal profile was at- 94 ℃ for 5 minutes followed by 39 cycles of 94 ℃ at 30 seconds, 55 ℃at 30 seconds and 72 ℃ for 1 minute with a final extension for 72 ℃ at 4 minutes. The COⅡ primer sequences were-Leu 5’- TCTAATATGGCAGATTAGTGCA -3’ and Lys 5’-ACTTGCTTTCAGTCATCTAATG-3’. Amplified PCR products were purified by Invitrogen PureLinkTMPCR purification kits. Purified products were then sequenced in both directions. The forward and reverse sequences of each sequence were checked and edited manually using BioEdit Sequence Alignment Editor Software.
2.4. Phylogenetic analysis
Multiple sequence alignment for COⅡ was performed using clustal X by method of Thompsonet al[14]. The final alignment was imported into MEGA version 5[15] and used to compute a neighbor-joining (NJ) phylogeny using Kimura two-parameter (K2P) model (Figure 1). Haplotype names were randomly assigned.An. albimanus(AAU92372) andAn. aquasalis(AAU92374) COⅡ sequences from Genebank were included in the analysis as outgroups. Tajima’s neutrality test[16], a test for the standard coalescent model was performed on the haplotypes using Arlequin 3.5.1.2.
2.5. Networks
Networks provide a way of representing more of the phylogenetic information present in a data set[17]. A statistical parsimony network tree was constructed using the software TCS version 1.21[18]. The program collapses sequence into haplotypes and calculates the frequencies of the haplotypes in the sample.
3. Results
From 25 selected study sites, 361 anopheline mosquitoes were morphologically identified. By ASPCR, only 76 specimens were identified asAn.minimus s.s.from 12 study areas. Specimens representing 9 sites were amplified for COⅡ markers. All species sequences were deposited in GenBank [accession numbers: JQ 046381 (seq 1), JQ 046382 (seq 2), JQ 046383 (seq 4), JQ 046386 (seq 3), JQ 046387 (seq 5), JQ 046388 (seq 6), JQ 046389 (seq 7), JQ 046390 (seq 8) and JQ 046391(seq 9)]. The analysis was based on 546 bp of COⅡ sequence obtained from 9 individuals representing 9 sites. All theAn. minimus s.s.sequences were Adenine (A)-Thymine (T) rich (74.18% A and T bases). Mutations were identified at 6 of the 546 variable sites, all of which were transitions. Among these, five of the substitutions resulted in amino acid change: at position 14 (L→P) in Haplotype 8, at position 131 (M→T) in Haplotypes 4, 6, 7 and 9; atposition 133 (S→F) in Haplotype 3; at position 167 (L→P) in Haplotype 8 and at position 169 (E→G) in haplotypes 2, 3, 5 and 8. A single non synonymous substitution was observed in a single specimen at position 460 (T →C).
The neighbour joining (NJ) tree generated distinct clusters of haplotypes (Figure 1). The first cluster (A) contained 3 haplotypes from Piong, Manas and Karbi Anglong. 3 more haplotypes were found in the second cluster (B) which originated from Banderduwa, Bhalukpong and Kimin. The third cluster (C) samples were obtained from Kimin and Boko. The lcations have been plotted in a map (Figure 2).
Statistical parsimony network analysis of 9 population sites generated a bifurcating tree showing population diversion of the samples (Figure 3).
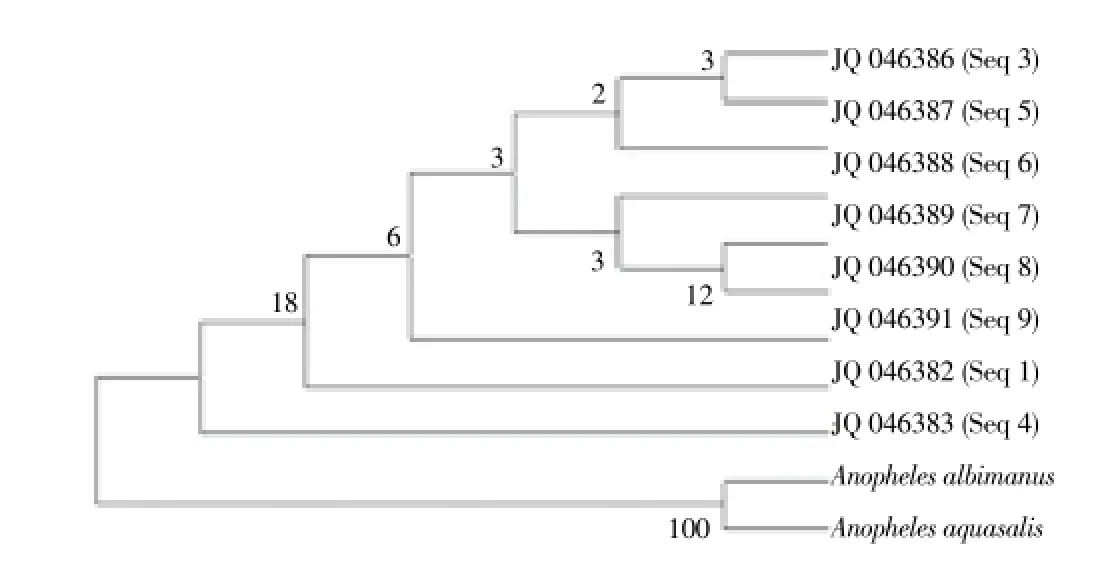
Figure 1. Evolutionary history using the Neighbor-Joining method and evolutionary distances using the Kimura 2-parameter method on an alignment of 368 positions in MEGA5.The reliability was tested by running 1 000 bootstrap replicates and the tree was rooted using a sequence from Anopheles albimanus (AAU92372) and Anopheles aqualis (AAU92374).

Figure 2. GIS map of North east India showing lcations of three clusters of haplotypes of An.minimus species.
Neutrality test statistics were found negative for all the 9 population sites. Tajima’s neutrality test on the nineAn. minimushaplotypes resulted in a D value of -0.410. This result was not significantly different from 0. Thus the null hypothesis, ie. the dataset being neutral, cannot be rejected. Fu’s FStest resulted in -8.865 which provide evidence of an excess number of alleles as would be expected from a recent population expansion or genetic hitch-hiking[19].

Figure 3. Genealogical relationships among haplotypes of COⅡgenes estimated by TCS.The ovals/ boxes indicate one of haplotype and the number within represents the type of haplotype. A unit branch represents one mutation. The empty circles indicate haplotypes that were not observed.
4. Discussion
The present distribution of genetic variation within a species happens due to its demographic history as well as the interaction between mutation, genetic drift and gene flow. Impact of Pleistoene glaciations in South East Asian region is yet to be studied. There is a need to explore factors that have shaped the current distributions of species in the region and the genetic variation within species.An.minimus s.shas been reported as one of major malaria vectors in NER. The present report is the first study of mitochondrial genetic variation analysis amongAn.minimus s.s.from this region of India.
Samples were collected from a total of 25 sites andAn.minimus s.s.were confirmed from only 12 sites that are reportedly malaria endemic lcoales. However, species representing 9 demographic sites were analyzed for gene diversification using COⅡgene. The newly sequenced mitochondrial genomes ofAn. minimusspecies were found to contain A-T rich (74.18% A and T bases) region. The A-T rich region in insects is known to contain regulatory sequences responsible for controlling replication and transcription of the mitochondrial genome[9].
Haplotype diversity and low nucleotide diversity werestudied by employing Tajima’s D test and Fu’s FStest. Tajima’s D test is based on the allele frequency distribution of segregating nucleotide sites. A positive value indicates a bias towards intermediate frequency alleles, while a negative value indicates a bias towards rare alleles, the latter being a signature of recent population expansion. Fu’s FStest is based on the distribution of alleles or haplotypes, and here too, negative values can indicate recent population growth. Both Tajima’s test and Fu’s FStest showed negative values. It has been shown that Fu’s FStest is more powerful than Tajima’s D, and this would explain the differences in significance for some populations. The overall negative values resulting from both tests indicate that there is an excess of rare mutations in the populations that could imply recent population expansion.Thus it can be expected that there is a recent population expansion or genetic hitch-hiking[19]. Alternatively, these values can result from balancing selection on a nearby lcous, although studies demonstrating direct or indirect selection (through hitchhiking) on the mitochondrial genome in natural populations are rare.
Network analysis of 9 haplotypes generated bifurcating tree that showed genetic variations among the sequences of different haplotypes that was also showed by mutations observed in 6 sequences.
The high genetic diversity/gene flow observed within the populations ofAn.minimusspecies has several implications for vector control. It can be suggested that mosquitoes could provide useful model systems for phylogeographic studies in Southeast Asia. A better taxonomic and ecological understanding of prevailing mosquito species is necessary in the region because mosquitoes are important as disease vectors. The vector status also means that there is a need to understand present-day distributions and population structure as an aid to control, for example in the management of insecticide resistance.
Conflict of interest statement
We declare that we have no conflict of interest.
Acknowledgements
The authors would like to thank Mr. C.K. Sharma, Mr. P. Doloi and Mr. L. Borah for rendering Field assistance. Financial assistance for this work was provided by the Indian Council of Medical Research (ICMR), Delhi, India. We would also like to acknowledge Mr. M.J. Nath for providing GIS map of Northeast India. Rashmee Topno and Pritom Chowdhury are supported by ICMR, Senior Research Fellowship.
[1] WHO. Malaria control today: Current WHO recommendations. RBM Department, Geneva: WHO; 2005.
[2] Dash AP, Valecha N, Anvikar AR, Kumar A. Malaria in India: Challenges and opportunities. J Biosci 2008; 33: 583-592.
[3] Das A, Anvikar AR, Cator LJ, Dhiman RC, Eapen A, Mishra N, et al. Malaria in India: The center for the study of complex malaria in India. Acta Trop 2012; 12: 267-273.
[4] Malaria situation in SEAR countries. World Health Organization. 2010. [Online] Available from: http://www.searo.who.int/en/ section10/section21/section340_4021.htm.
[5] Malaria situation in India. National Vector Borne Disease Control Programme. [Online] Available from: http://www.nvbdcp.gov.in/ Doc/mal-situation-June12.pdf
[6] Dev V, Phookan S, Sharma VP, Anand SP. Physiographic and entomologic risk factors of malaria in Assam, India. Am J Trop Med Hyg 2004; 71: 451-456.
[7] Dutta P, Baruah BD. Incrimination of Anopheles minimus theobald as a vector of malaria in Arunachal Pradesh. Indian J Malariol 1987; 24: 59-162.
[8] Dutta P, Mahanta J. Incrimination of Anopheles minimus as a vector of malaria in Karbi Anglong district of Assam. Indian J Malariol 1995; 32: 129-131.
[9] Krzywinskia J, lib C, Morrisa M, Connd JE, Limaf JB, Povoag MM, et al. Analysis of the evolutionary forces shaping mitochondrial genomes of a Neotropical malaria vector complex. Mol Phylogenet Evol 2011; 58: 469-477.
[10] Das BP, Rajagopal, R, Akiyama, J. Pictorial key to the species of Indian anopheline mosquitoes. Zoology 1990; 2: 131-162.
[11] Mohanty A, Kar P, Mishra K, Singh DV, Mohapatra N, Kar SK, et al. Multiplex PCR assay for the detection of Anopheles fluviatilis species complex, human host preference and Plasmodium falciparum sporozoite presence using a unique mosquito prcoessing method. Am J Trop Med Hyg 2007; 76: 837-843.
[12] Phuc HK, Ball AJ, Son L, Hanh NV, Tu ND, Lien NG, et al. Multiplex PCR assay for malaria vector Anopheles minimus and four related species in the Myzomyia series from Southeast Asia. Med Vet Entomol 2003; 17: 423-428.
[13] Morgan K, O’Loughlin SM, Yik FM, Linton YM, Somboon P, Min S, et al. Molecular phylogenetics and biogeography of the Neocellia series of Anopheles mosquitoes in the Oriental Region. Mol Phylogenet Evol 2009; 52: 588-560.
[14] Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. The Clustal ×windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 1997; 24: 4876-4882.
[15] Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 2011; 28: 2731-2739.
[16] Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 1989; 123: 585-595.
[17] Posada D, Crandall KA. Intraspecific gene genealogies: trees grafting into networks. Trends Ecol Evol 2001; 16: 37-45.
[18] Clement M, Posada D, Crandall K. TCS: a computer program to estimate gene genealogies. Mol Ecol 2000; 9: 1657-1660.
[19]Fu YX. Statistical tests of neutrality of mutations against population growth, hitch-hiking, and background selection. Genetics 1997; 147: 915-925.
ment heading
10.1016/S1995-7645(14)60168-1
*Corresponding author: Dr Prafulla Dutta, Scientist F, RMRC, ICMR (NE Region), Dibrugarh-786001, Post Box-105, Assam, India.
Tel: (+91)-(0373) - 2381494, 2381506
Fax : (+91) - (0373) - 2381748
E-mail: duttaprafulla@yahoo.com
 Asian Pacific Journal of Tropical Medicine2014年12期
Asian Pacific Journal of Tropical Medicine2014年12期
- Asian Pacific Journal of Tropical Medicine的其它文章
- Chikungunya virus, epidemiology, clinics and phylogenesis: A review
- Induction of deletion mutation on ompR gene of Salmonella enterica serovar Typhi isolates from asymptomatic typhoid carriers to evolve attenuated strains for vaccine development
- Evaluation of protective effect of IL-22 and IL-12 on cutaneous leishmaniasis in BALB/c mice
- Immunogenic potential and protective efficacy of formalin inactivated circulating Indian strain of West Nile virus
- Fumigant and repellent properties of sesquiterpene-rich essential oil from Teucrium polium subsp. capitatum (L.)
- Antioxidant activity and free radical scavenging activities of Streptomyces sp. strain MJM 10778