
过氧化物酶体增殖物激活受体激动剂类药物的致癌性和致癌机制研究进展
2014-03-23 09:12:37邢立国吴英良
中国药理学与毒理学杂志 2014年3期
邢立国,吴英良
(1.沈阳化工研究院安全评价中心国家沈阳新药安全评价研究中心,辽宁沈阳 110021;2.沈阳药科大学生命科学与生物制药学院,辽宁沈阳 110016)
过氧化物酶体增殖物激活受体激动剂类药物的致癌性和致癌机制研究进展
邢立国1,2,吴英良2
(1.沈阳化工研究院安全评价中心国家沈阳新药安全评价研究中心,辽宁沈阳 110021;2.沈阳药科大学生命科学与生物制药学院,辽宁沈阳 110016)
过氧化物酶体增殖物激活受体(PPAR)是一类由配体激活的核转录因子,参与糖类和脂类代谢、炎症反应、细胞生长和分化等过程,以其为靶点的降脂类及抗糖尿病药物已经被开发。PPAR激动剂对动物有致癌性,如一些贝特类PPARα激动剂、噻唑烷二酮类PPARγ激动剂、开发的PPARα/γ双重激动剂和PPARδ激动剂均可使实验动物发生肿瘤。PPARα激动剂的致癌机制同PPARα受体有关,激活受体调节代谢产生脂类异常,也引起过氧化物酶体氧化酶活性增加,产生活性氧导致DNA的损伤。枯否细胞通过NADPH氧化酶产生活性氧促进肝细胞增殖,抑制凋亡。PPARγ激动剂的致癌性与结石形成有关。PPAR激动剂是否对人具有致癌性尚未证实,临床应用仍有致癌风险。本文主要综述PPAR激动剂致癌性和致癌机制研究进展,希望对该类药物的开发有所帮助。
过氧化物酶体增殖物激活受体激动剂;毒性作用;致癌物
DO l:10.3867/j.issn.1000-3002.2014.03.025
过氧化物酶体增殖物激活受体(peroxisom e p ro liferators activated receptors,PPAR)属于非甾体类核受体超家族,是一类由配体激活的核转录因子。根据其结构不同,可分为PPARα、PPARβ(或PPARδ)及PPARγ3种亚型。PPAR参与调节脂质代谢、脂肪生成、胰岛素敏感性、炎症反应、细胞生长和分化等重要生化反应及生物调节过程。一系列代谢综合征,如糖尿病、肥胖、高脂血症、高血压病、动脉粥样硬化症等均与PPAR有关,PPAR成为当前人类代谢相关疾病药物的重要靶标。PPAR激动剂类药物在近年得到广泛的开发,但因最早发现的PPARα激动剂在实验动物上的致癌性问题,使该类药物的致癌性成为安全性评价的焦点。
1 PPAR激动剂类药物的致癌性
PPARα激动剂能够促进脂类代谢和增加高密度脂蛋白的合成,贝特类(fibrates)降脂药氯贝特和苯扎贝特等是最先发现的PPARα人工合成配体,其他如吉非贝齐(gem fibrozil)、非诺贝特(fenofibrate)和氯贝丁酯(clofibrate)等已作为降脂药用于临床。动物实验(主要是大鼠和小鼠长期致癌实验)中,发现贝特类药物都具有致肝癌作用,但因一直没有临床证据,所以部分药品临床还在使用。
噻唑烷二酮类(thiazolidinediones,TZD)PPARγ激动剂具有胰岛素增敏作用,是典型的抗糖尿病药物,其中曲格列酮(trog litazone)因严重肝毒性已于2000年退出市场,罗格列酮(rosiglitazone)和吡格列酮(pioglitazone)曾是使用最广泛的药物。吡格列酮在动物实验中出现膀胱肿瘤[1],流行病学调查发现膀胱癌风险[2-3],尽管结论仍然存在争议[4-5],2011年美国食品药物管理局(FDA)和欧洲医药管理局(EMA)发出警告,法国和德国则从市场撤销该药物[6]。罗格列酮虽没有肝毒性,但因其心血管风险[7],2011年被EMA和美国FDA从市场上暂停和限用,致癌性不是考虑因素。高剂量罗格列酮在大鼠致癌实验有发生脂肪瘤的可能,同时罗格列酮本身具有一定的PPARα激动性,因此,有致肝癌的可能性[8]。PPAR药物在临床应用时间较长,是治疗糖尿病的主要药物,对致癌性还要谨慎评估[9]。
PPARγ激动剂作用于脂肪组织,促使葡萄糖向脂肪组织转运,这可能增加患者体重,而PPARα激动剂可以促进脂类的氧化代谢,与PPARγ激动剂有协同作用,因此PPARα/γ双重激动剂能较好地解决PPARγ激动剂的增加体重的副作用,成为近年该类药物的开发主流[10]。目前PPARα/γ双重激动剂开发面临的一个重要安全问题就是致癌性的困扰。因为该类化合物同时具有PPARα的活性,其肝致癌潜力倍受关注。虽然很多PPARα/γ双重激动剂的临床前研究结果令人满意,但临床Ⅲ期试验中却因为不良反应和安全性问题而不得不终止,其中致癌潜力是一个重要因素,如治疗糖尿病候选药JTT-501因致腺体瘤于2002年中止,MK-767因致血管肉瘤于2004终止[11],拉格列扎(ragag litazar)因致泌尿道上皮癌于2003年Ⅲ期临床后中止,莫格列扎(muraglitazar)2006年因致膀胱癌风险被美国FDA暂停[8]。2004年,美国FDA根据处于开发阶段的6个PPARα+γ和5个PPARγ化合物的致癌实验结果认为,PPAR激动剂是多种属、多品系、多性别及多位点的致癌物[12]。Oleksiew icz等[13]总结了PPARα激动药或化学品、PPARγ激动剂和PPARα/γ激动剂的致癌性结果,发现不同的激动剂致癌性存在差异,PPARα激动剂动物主要产生肝肿瘤,PPARγ激动剂产生血管瘤和脂肪肉瘤等,PPARα/γ激动剂产生肿瘤同PPARγ相似,但发生率更高,两者肿瘤发生率更广泛。如表1所示。PPAR激动剂是非遗传致癌物,多位点发生肿瘤,与PPAR的组织分布有一定的相关性。
一些研究证明,PPARγ激动剂对各种肿瘤细胞均具有抑制作用,有望开发为抗肿瘤药物,但在临床前评价却发现该类化合物具有致癌性。虽然有证据显示曲格列酮能够抑制啮齿类动物结直肠肿瘤的生成,但后来发现其不仅增强有结肠肿瘤生成趋势的突变鼠的癌变,也诱发了没有基因修饰的正常鼠的结肠肿瘤[14]。PPARγ激动剂的抑癌和促癌机制尚不清楚,其致癌性问题限制了药物的开发。
PPARδ激动剂类药物开发较晚,致癌性研究有限,且报道结果存在争议[15-16]。PPARδ激动剂GW 501516在转基因小鼠模型中产生小肠息肉[17],在大鼠致癌实验中多组织产生肿瘤,如肝细胞癌、膀胱癌、甲状腺癌和雌鼠子宫内膜癌等[18]。PPARδ激动剂产生广泛的肿瘤,可能是实验剂量过高脱靶效应导致的,据报道,低剂量PPARδ激动剂可产生抑制肿瘤的效应[19-20]。随着PPARα/δ,PPARγ/δ和PPARα/δ/γ双激动剂和泛激动剂的开发,对该类药物的致癌性应该给予充分重视。
2 PPAR激动剂致癌的可能机制
自1979年首次发现降血脂药物氯贝丁酯的致癌性以来,过氧化物酶体增殖剂药物的致癌性被广泛研究。对该类药物致癌性的研究直接导致了PPAR的发现,加深了肿瘤发生的认识,建立的受体激活的非遗传致癌途径,这是对传统遗传致癌理论的一大进步。属于过氧化物酶体增殖物(peroxisom e pro liferator,PP)的化合物种类很多,不仅包括开发的药物、还包括工业用增塑剂,如邻苯二甲酸(2-乙基己基)酯〔bis(2-ethylhexyl)orthophthalate,DEHP〕和环境污染物等。对PPAR激动剂致癌性的理解很大程度上来源于对工业增塑剂等的研究。PPARα激动剂致癌机制不断被阐明,为PPARγ和PPARδ的研究提供了启示。
2.1 受体依赖致癌
1990年Issemann等[21]首次鉴定了PPAR,通过小鼠基因敲除实验证明PPARα是肝肿瘤发生的必要条件。一般认为,大鼠肝过氧化物酶体增生同PPARα配体处理相关,是肿瘤发生的早期事件,但近年也有研究发现,过氧化物酶增生同肿瘤的发生没有联系,说明PPARα配体致癌的复杂性[22]。
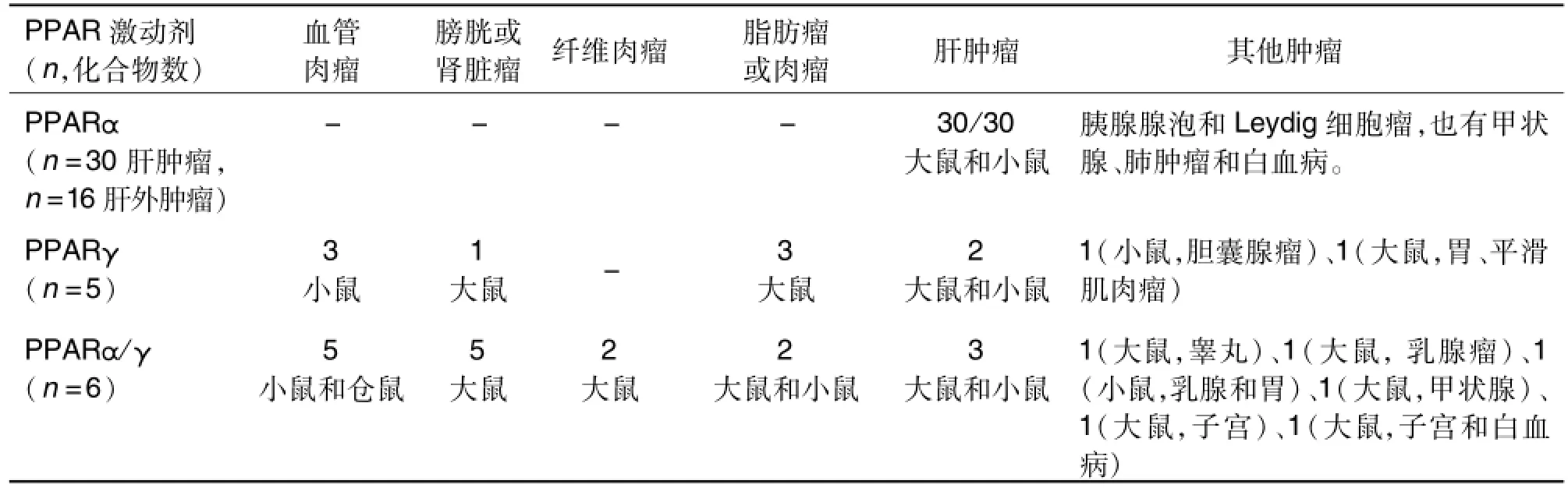
表1 过氧化物酶体增殖物激活受体(PPAR)激动剂导致大鼠、小鼠和仓鼠发生的肿瘤[13]
PPAR的主要功能是调节基因的转录。首先与配体结合而被激活,然后与9-顺视黄酸类受体/维A酸受体X或糖皮质激素受体形成异二聚体,再与靶基因的启动子上游的PP反应元件结合而使靶基因活化,从而调节靶基因的转录表达。PPAR调控的许多靶基因与脂质代谢有关,但是否受体直接调控基因决定肿瘤的发生,还是有更复杂的机制仍不清楚,详细过程还要进行深入研究。
2.2 脂类代谢异常和炎性反应
PPARα激动剂影响脂肪酸的摄取、结合、氧化以及脂蛋白的装配和转运,提高脂质分解代谢。DEHP活化的基因主要包括:酰基辅酶A合成酶、酰基辅酶A氧化酶(acyl-CoA oxidase,ACO)、中链酰基辅酶A脱氢酶、细胞色素P450 4A6以及肝脂肪酸结合蛋白、脂肪酸转运蛋白、脂肪酸转位酶以及载脂蛋白AⅠ,AⅡ和CⅢ等基因的转录和活化。因此,PPAR在脂质代谢(线粒体和过氧化物酶体β氧化,微粒体羟化)中占有重要地位,影响体内脂质平衡。脂肪的许多代谢产物如胆固醇、胆汁酸、花生四烯酸衍生物等同肿瘤的发生相关。
PPARα激动剂诱导过氧化物酶体增生,而过氧化物酶体β氧化与胆汁酸合成途径相关[23]。细胞色素P450 7A1催化胆固醇向胆汁酸转变的第一步反应,是胆汁酸合成的限速酶,其表达水平的高低反映胆汁酸合成的快慢。研究表明,应用DEHP后细胞色素P450 7A1 mRNA表达升高,同时粪胆汁酸排出量也增加,表明DEHP激活PPARα促进了肝胆汁酸的合成。降脂药物同时降低胆固醇,胆固醇向胆汁酸的转化是一条重要途径。胆汁酸淤积导致肝肿瘤,在肠道经微生物修饰促进结肠癌的发生[24]。
PPARγ激动剂吡格列酮致大鼠膀胱癌,可能是由于尿道和膀胱中形成结石,诱导细胞毒性,导致浅层上皮炎性坏死,细胞持续增殖,最终发生肿瘤[25]。PPARα/γ双重激动剂莫格列扎也可能通过尿路结石途径导致细胞炎性坏死,进而演进发生膀胱肿瘤[26]。
2.3 氧化应激
氧化应激失衡可能是PP诱导肿瘤的一个重要途径。过氧化物体是线粒体外物质代谢的细胞器,也是活性氧(reactive oxygen species,ROS)产生的重要细胞器。酶体物质氧化产生H2O2,被过氧化氢酶分解为H2O和O2循环使用。在大鼠肝过氧化物酶体产生35%的H2O2,占总氧耗的20%。在金属催化下H2O2易转化为·OH,羟基自由基损伤细胞。过氧化物酶体中黄嘌呤氧化酶氧化反应产生的超氧阴离子(O÷2)具有更强的氧化性,一氧化氮合酶催化精氨酸产生NO,NO能同O÷2反应生成过氧亚硝基阴离子(ONOO-)。当过氧化物酶体产生ROS超过抗氧化能力时,可能导致肿瘤的发生[27]。PPARα激动剂处理可引起肝过氧化物酶体大量生成,过氧化物酶体中介导脂肪β氧化的ACO活性增加,但同时使H2O2破坏的过氧化氢酶并没有增加,这种氧化酶/过氧化氢酶比例的不平衡可导致氧化物增多[28]。不能被降解的H2O2可能从过氧化物酶体中泄漏,在体内可形成具有高度反应性能的羟基,并对DNA产生氧化损伤。ROS能与核酸形成加合物,如8-羟基鸟苷(8-OHdG),在DNA复制期间产生突变。在给予各种PP的大鼠中,可见以8-OHdG为表现形式的DNA损伤。同时使用新型的碱基切除修复系统也观察到在PP处理的大鼠、小鼠肝的核酸氧化损伤现象[29]。
但目前对PP通过DNA损伤途径致癌的观点存在争议,如DNA加合物8-OHdG测量通常使用整个动物肝提取物,因此无法区分它的来源,线粒体也能产生该加合物。有研究证明,从肝细胞核提取的DNA,8-OHdG同对照组没有差异;也有研究证明在PP处理的细胞核中8-OHdG含量增加,但达不到同肿瘤发生相关的程度[30]。此外,在氯贝丁酯处理的动物体内并没有H2O2的增加,也没有DNA链的断裂;在ACO基因敲除的小鼠实验中也证明ACO不是肿瘤发生必须的[31]。
NADPH氧化酶最初发现存在于吞噬细胞的质膜,能催化底物NADH/NADPH产生O÷2和过氧化氢(H2O2)等ROS。枯否细胞是肝中的巨噬细胞,可能参与PPARα激动剂介导的肿瘤发生。枯否细胞PPAR激动后通过NADPH氧化酶氧化反应产生ROS,ROS通过NF-κB途径调控肿瘤坏死因子α(tumor necrosis factor-a lpha,TNF-α)、白细胞介素1等细胞因子表达,细胞因子起到类似丝裂原的活性促进肝细胞的增殖,并抑制细胞的凋亡[32-33]。进一步的研究认为,枯否细胞主要在急性阶段起作用,对药物的长期暴露影响不大[34]。枯否细胞在肿瘤发生中的作用,特别是和其他细胞间的协同作用值得进一步研究。
线粒体是产生ROS的主要场所,细胞内氧化还原电位改变增加ROS产生。研究发现,PPARα和PPARγ激动剂均能抑制线粒体呼吸链复合物Ⅰ,抑制电子从NADH传递给辅酶Q,从而使电子在呼吸链上传递受阻而外漏增加,进而与O2结合使细胞内ROS的产生增加[35]。然而DEHP处理后线粒体解偶联蛋白过量表达,由于解偶联蛋白具有很高的质子转运活性,能使质子直接进入基质而不参与ATP的合成,结果导致储存在质子电化学势能中的自由能以热能形式被消耗,降低了质子电化学势能梯度,从而也抑制ROS的生成,这同前述报道相矛盾。线粒体在PPARγ激动剂介导的ROS产生过程中的作用仍不清晰。
ROS不仅能够诱导细胞凋亡,也是细胞增殖的信号转导分子。细胞增殖或凋亡的平衡主要同ROS的浓度有关,高浓度诱导凋亡,而低浓度促进增殖。
2.4 促进细胞增殖,抑制细胞凋亡
促进细胞增殖和抑制细胞凋亡是PP诱导肝肿瘤的主要机制之一。许多体外和体内研究都表明,PP处理后肝细胞的DNA合成增加。肝细胞生长改变是肝癌发生的前提,PP致肝细胞增殖的作用包括急性、慢性和癌前病变。大鼠经口给予PP后,肝重量急剧增加造成肝肿大,主要由于肝细胞增殖。在诱导肝肿大过程中,也发生肝细胞凋亡,肝重量增加是可逆的。虽然短期的肝肿大和细胞的增殖程度不能预测肿瘤的发生,但确是致癌性的PP暴露后都会发生的共同特征。急性期主要是炎性介质增加,肝细胞有丝分裂原肿瘤坏死因子(TNF)-α发挥作用。大鼠长期暴露于PP肝细胞增殖也被观察到,很可能是肝细胞增殖被细胞凋亡平衡,使肝质量达到了平台。慢性细胞增殖的持续增长是预测致癌性的很好指标。细胞增殖作用诱发肝癌的表现是癌前病灶的肝细胞选择性生长。肝细胞病灶有明显核仁,核质比增加,嗜碱性细胞质,称为嗜碱性灶。病灶和腺瘤细胞凋亡水平增加,但因为细胞复制比率高于细胞凋亡,细胞总数增加。诱发癌变需要PP的持续暴露,如果撤消暴露则细胞的增殖降低凋亡增加,最终病灶退化[36]。
虽然在病理组织学和DNA合成分析中发现肝细胞持续增生,但增殖发生的细胞生物学过程仍不清楚。细胞增殖增加基因突变的频率,可能发生癌基因的激活和抑癌基因的突变,通过持续的增殖,突变克隆选择保留,细胞最终发展成肿瘤。
抑制凋亡途径是PPAR致癌的另一个机制。通过凋亡清除启动细胞是阻止恶性转化的一种防御机制。PP除了增加肝细胞的增殖,还可减少正常细胞和启动细胞群的凋亡,有利于诱导肝癌的形成。凋亡抑制的分子机制并不清楚,活化的PPAR被证明是对细胞过氧化物酶体凋亡抑制必要的,PP给予PPAR基因敲除小鼠细胞凋亡不能被抑制[37]。作为PPARα的激活剂,大多数PP在实验中均可抑制细胞凋亡,该结果与在高浓度TNF-α下培养啮齿动物的肝细胞现象类似[38]。TNF-α在PPARα敲除的小鼠仍可抑制细胞凋亡,表明TNF-α可能是PP或PPARα抑制细胞凋亡的下游效应分子[37]。PP诱导的肝细胞的凋亡抑制还与信号转导密切相关,Barger等[39]研究显示,当PP以p38 MAPK依赖的方式激活时,PPARα靶基因的转录活性明显加强,而且PPARα中配体非依赖性的转录激活区域,即转录激活功能区1功能激活域中含有MAPK的位点,因而PPARα的激活取决于p38 MAPK诱导的磷酸化。另外,PP导致的氧化抑制在激活MAPK激酶途径中也起一定的作用,已证明p38 MAPK与氧化应激相联系。老年大鼠对PPARα激活剂的抗凋亡作用非常敏感,此作用与高水平表达的BcL-2相对应,PPARα激活剂WY-14643还可减弱凋亡因子,如Bax、胱天蛋白酶和Fas的水平[40]。
3 人类致癌的可能性
PP类物质作用具有明显的种属差异。大量研究表明,PP能诱导大鼠、小鼠肝肿瘤,但对灵长类动物几种PP类药物的长期慢性染毒并未诱导出肝肿瘤。使用一些标志性过氧化物酶的活性作为指标进行短期实验表明,大鼠和小鼠对PP类物质的作用最为敏感,豚鼠与犬的反应不明显或反应微弱,灵长类动物基本无反应。使用大鼠、小鼠、豚鼠、灵长类动物肝细胞原代培养进行体外实验,PP染毒得出的结果与体内实验一致。使用人原代培养肝细胞的PP类物质染毒,并未出现过氧化物酶体增殖以及过氧化物酶功能增强。这种现象可能是由于PPARα在不同属的表达存在差异,其在人类肝的表达水平只相当于大鼠、小鼠肝的1%~10%[41],动物和人的PPAR受体激动的活性也不相同。人和大鼠的ACO基因启动子不同,人的ACO基因不含过氧化物酶体增殖物反应元件,不能对PPARα的诱导产生应答[42]。人类细胞对PP物质的基因反应不同,可能是影响致癌性的因素[43]。尽管流行病学调查表明,PP类降血脂药物临床使用了近30年没有发现致癌倾向,但因为PP类物质在临床上存在和大鼠相似的药效学反应,人类长期应用PP类药物存在的致癌性风险应该重视。
药物对人的安全性评估建立在致癌作用机制之上,前文所述的几种假说当前还有很多争议,某些环节还缺乏证据,因此,在临床使用上仍需谨慎。针对PPAR药物的安全性评价非常严格,很多药物因为致癌风险在Ⅲ期临床阶段终止研究,上市的药物也有安全风险。
4 展望
对PPAR激动剂的致癌性研究了近30年,大致了解其致癌机制。受体依赖的致癌性是主要途径,激动剂通过受体激活调控下游基因的转录,间接刺激细胞增殖和抑制细胞凋亡,长期作用最终促进肿瘤的发生。但详细的发生过程仍不清楚,致癌关键事件还存在很多争议。如促进细胞增殖过程,大量研究认为TNF-α发挥作用,但转基因动物证明TNF-α不是PP致癌必须的。致癌过程还有许多问题没有解决,未来的研究应该弄清肿瘤细胞的发生来源,发生的细胞学过程,如肿瘤干细胞是否发挥作用;研究凋亡抑制的机制,PPAR调控的线粒体能量代谢内质网应激在凋亡抑制和肿瘤发生中可能起重要作用[44];深入研究肿瘤发生关键过程,如信号途径、表观学改变、m icroRNA变化等,如已有文献证明m icroRNA let-7c在PPARα激动剂的致癌中发挥作用[45];不同PPAR亚型激动剂致癌机制的差异等。
PPAR受体参与重要的生理过程,针对其开发的激动剂类药物存在高度的致癌性风险,药物开发需要对其功能进行深入了解和把握。PPAR激动剂类药物致癌机制的研究有助于PPAR激动剂药物研发,设计时尽可能避免致癌因素,以降低研发失败的风险,最终提高临床应用的安全性。
[1] Cohen SM.Effects of PPARgamma and combined agonists on the urinary tract of rats and other species[J].Toxico l Sci,2005,87(2):322-327.
[2] Azoulay L,Yin H,Filion KB,Assayag J,Majdan A,Pollak MN,et al.The use of pioglitazone and the risk o fb ladder cancer in peop le w ith type 2 diabe tes:nested case-control study[J].BMJ,2012,344:e3645.
[3] Hillaire-Buys D,Faillie JL,Montastruc JL.Pioglitazone and bladder cance r[J].Lancet,2011,378(9802):1543-1544.
[4] Tseng CH.Pioglitazone and bladder cancer:a population-based study of Taiwanese[J].Diabetes Care,2012,35(2):278-280.
[5] Song SO,Kim KJ,Lee BW,Kang ES,Cha BS,Lee HC.The risk of bladde r cancer in Korean diabetic subjects trea ted w ith pioglitazone[J].Diabetes Metab J,2012,36(5):371-378.
[6] Ba rbalat Y,Dombrovskiy VY,Weiss RE.Association between pioglitazone and urothelial bladder cancer[J].Urology,2012,80(1):1-4.
[7] Shukla R,Kalra S.Pioglitazone:Indian perspective[J].Indian J Endocrino lMe tab,2011,15(4):294-297.
[8] Rubenstrunk A,Hanf R,Hum DW,Fruchart JC,Staels B.Sa fety issues and prospects for future generations of PPAR modulators[J].Biochim Biophys Acta,2007,1771(8):1065-1081.
[9] Cariou B,Charbonnel B,Staels B.Thiazolidinediones and PPARγagonists:time for a reassessmen t[J].Trends Endocrinol Metab,2012,23(5):205-215.
[10] Luo WY,Liu YX.Problem s and prospects o f PPAR dual/pan agonists[J].Chin J New Drugs(中国新药杂志),2008,17(4):279-282,288.
[11] Balakumar P,Rose M,Ganti SS,Krishan P,Singh M.PPAR dual agonists:are they opening Pandora′s Box?[J].Pharmacol Res,2007,56(2):91-98.
[12] El-Hage J.Preclinical and clinical safety assesmen ts for PPAR agonists[B/OL].(2004)[2014-5-22].h ttp://www.fda.gov/downloads/AboutFDA/CentersOffices/OfficeofMedicalProductsand-Tobacco/CDER/UCM119070.ppt
[13] O leksiewicz MB,Southgate J,Iversen L,Egerod FL.Rat urinary bladder carcinogenesis by dua lac ting PPARalpha+gamma agonists[J].PPAR Res,2008,2008:103167.
[14] Yang K,Fan KH,Lam precht SA,Edelmann W,Kopelovich L,Kucherlapati R,et al.Peroxisome proliferator-activated receptor gamma agonist troglitazone induces colon tumors in normal C57BL/6J m ice and enhances colonic carcinogenesis in Apc1638N/+M lh1+/-double m utantm ice[J].Int J Cancer,2005,116(4):495-499.
[15] Mackenzie LS,Lione L.Harnessing the benefits o f PPARβ/δagonists[J].Life Sci,2013,93(25-26):963-967.
[16] Peters JM,Shah YM,Gonzalez FJ.The role o f pe roxisome p rolife rator-activated receptors in carcinogenesis and chemoprevention[J].Nat RevCancer,2012,12(3):181-195.
[17] Gupta RA,Wang D,Katkuri S,Wang H,Dey SK,DuBois RN.Activation of nuc lear hormone receptor peroxisome proliferator-activated receptordelta accelerates intestinal adenoma grow th[J].Nat Med,2004,10(3):245-247.
[18] Geiger LE,WSD,Lew is DJ,Brennan C,Liu KC,Newsholme SJ.Rat carcinogenicity study w ith GW501516,a PPAR de lta agonist[J/OL].Toxico logist,2009,108:895.http://www.toxicology.org/AI/Pub/Tox/2009 Tox.pdf
[19] Marin HE,Peraza MA,Billin AN,W illson TM,Ward JM,Kennett MJ,et al.Ligand activation of peroxisome proliferator-activated recep tor be ta inhibits co lon carcinogenesis[J].Cancer Res,2006,66(8):4394-4401.
[20] Hollingshead HE,Borland MG,Billin AN,Willson TM,Gonzalez FJ,Peters JM.Ligand activation of peroxisome proliferator-activated receptor-beta/delta(PPARbeta/delta)and inhibition of cyclooxygenase 2(COX2)attenuate colon carcinogenesis through independent signa ling mechanism s[J].Carcinogenesis,2008,29(1):169-176.
[21] Issemann I,Green S.Activation of a member of the steroid hormone receptor superfam ily by peroxisome p roliferators[J].Nature,1990,347(6294):645-650.
[22] Youssef JA,Badr MZ.Aging and enhanced hepatocarcinogenicity by pe roxisome proliferator-activated receptor alpha agonists[J].Ageing Res Rev,2005,4(1):103-118.
[23] Ferdinandusse S,Houten SM.Peroxisomes and bile acid biosynthesis[J].Biochim Biophys Acta,2006,1763(12):1427-1440.
[24] Debruyne PR,Bruyneel EA,Li X,Zimber A,Gespach C,Mareel MM.The role of bile acids in carcinogenesis[J].Mutat Res,2001,480-481:359-369.
[25] SuzukiS,Arnold LL,Pennington KL,Kakiuchi-Kiyota S,Wei M,Wanibuchi H,et al.Effects of pioglitazone,a peroxisome proliferator-activated receptor gamma agonist,on the urine and urothelium of the rat[J].Toxicol Sci,2010,113(2):349-357.
[26] Tannehill-G regg SH,Sanderson TP,Minnema D,Voelker R,Ulland B,Cohen SM,et al.Rodent carcinogenicity profile of the antidiabetic dualPPAR alpha and gamma agonistmurag litazar[J].Toxico l Sci,2007,98(1):258-270.
[27] Schrader M,Fahim i HD.Peroxisomes and oxidative stress[J].Biochim Biophys Acta,2006,1763(12):1755-1766.
[28] Nilakantan V,Spear BT,G lauert HP.Effectof the peroxisome p roliferator ciprofibrate on lipid peroxidation and 8-hydroxydeoxyguanosine formation in transgenic m ice w ith elevated hepatic catalase activity[J].Free Radic Biol Med,1998,24(9):1430-1436.
[29] Rusyn I,Denissenko MF,Wong VA,Butterworth BE,Cunningham ML,Upton PB,et al.Expression of base excision repair enzymes in rat and mouse liver is induced by peroxisome proliferators and is dependent upon carcinogenic potency[J].Ca rcinogenesis,2000,21(12):2141-2145.
[30] Bosgra S,Mennes W,Seinen W.Proceedings in uncovering the mechanism behind peroxisome proliferator-induced hepatocarcinogenesis[J].Toxicology,2005,206(3):309-323.
[31] Fan CY,Pan J,Usuda N,YeldandiAV,Rao MS,Reddy JK.Steatohepatitis,spontaneous peroxisom e pro lifera tion and liver tumors in m ice lacking pe roxisomal fatty acyl-CoA oxidase.Im p lications fo r peroxisome proliferato r-ac tivated receptor alpha na tural ligand m etabolism[J].J Biol Chem,1998,273(25):15639-15645.
[32] Rose ML,Rusyn I,Bojes HK,Belyea J,Cattley RC,Thurman RG.Ro le of Kupffer cells and oxidants in signaling peroxisome proliferator-induced hepatocyte proliferation[J].Mu tatRes,2000,448(2):179-192.
[33] G lauert HP,Calfee-Mason K,Li Y,Nilakantan V,Tw aroskiML,Tharappel J,et al.The role of NF-kappaB in PPARalpha-mediated hepatocarcinogenesis[J].PPAR Res,2008,2008:286249.
[34] Woods CG,Burns AM,Bradford BU,Ross PK,Kosyk O,Swenberg JA,et al.WY-14 643 induced ce ll proliferation and oxidative stress in mouse liver are independen t of NADPH oxidase[J].Toxicol Sci,2007,98(2):366-374.
[35] Scatena R,BottoniP,Giardina B.Mitochondria,PPARs,and cancer:Is receptor-independent action of PPAR agonists a key?[J].PPAR Res,2008,2008:256251.
[36] Corton JC,Lapinskas PJ,Gonzalez FJ.Central role of PPARa lpha in the m echanism of ac tion of hepa tocarcinogenic peroxisome proliferators[J].Mutat Res,2000,448(2):139-151.
[37] Hasma ll SC,James NH,Macdonald N,Gonza lez FJ,Peters JM,Roberts RA.Suppression o f m ouse hepatocyte apoptosis by peroxisome proliferato rs:role of PPARalpha and TNFalpha[J].Mutat Res,2000,448(2):193-200.
[38] Rolfe M,James NH,Roberts RA.Tumour necrosis fac tor alpha(TNF alpha)suppresses apoptosis and induces DNA synthesis in rodent hepatocytes:a mediator o f the hepatocarcinogenicity of peroxisome proliferators?[J].Carcinogenesis,1997,18(11):2277-2280.
[39] Barger PM,Browning AC,Garner AN,Kelly DP.p38 m itogen-activated protein kinase activates peroxisome proliferato r-ac tivated receptor alpha:a potential role in the cardiac metabolic stress response[J].J BiolChem,2001,276(48):44495-44501.
[40] Youssef J,Badr M.Enhanced hepatocarcinogenicity due to agonists of peroxisom e proliferator-activated receptors in senescent rats:role of peroxisome proliferation,cellp roliferation,and apoptosis[J].SciWorld J,2002,2:1491-1500.
[41] Doull J,Cattley R,Elcombe C,Lake BG,Swenberg J,W ilkinson C,et al.A cancer risk assessment of di(2-ethylhexyl)phthalate:application of the new U.S. EPA Risk Assessment Guidelines[J].Regul Toxicol Pharmacol,1999,29(3):327-357.
[42] Roberts RA,James NH,Hasmall SC,Holden PR,Lambe K,Macdona ld N,et al.Apoptosis and proliferation in nongenotoxic carcinogenesis:species differences and ro le of PPARalpha[J].Toxicol Lett,2000,112-113:49-57.
[43] Kim S,Kiyosaw a N,Burgoon LD,Chang CC,Zacharewski TR.PPARα-mediated responses in human adult liver stem ce lls:In vivo/in vitro and cross-species com parisons[J].J Steroid Biochem Mol Biol,2013,138:236-247.
[44] Misra P,Reddy JK.Peroxisome proliferator-activated receptor-αactivation and excess energy bu rning in hepatocarcinogenesis[J].Biochim ie,2014,98:63-74.
[45] Gonza lez FJ,Shah YM.PPARalpha:mechanism of species differences and hepatoca rcinogenesis o f pe roxisome proliferators[J].Toxicology,2008,246(1):2-8.
Progress o f carcinogenesis and possib le mechanisms of peroxisome p ro liferato r-ac tivated recep to r agonists
XING Li-guo1,2,WU Ying-liang2
(1.Safety Evaluation Center of Shenyang Research Institute o f Chem ical Industry Ltd.,National Shenyang Center for Drug Sa fety Evaluation and Research,Shenyang 110021,China;2.Co llege of Life Science and Biopharm aceutica l,Shenyang Pharmaceutica l University,Shenyang 110016,China)
Peroxisome proliferator-activated receptors(PPARs)are ligand-activated nuclear transcription factors,playing an important role in the regulation of glucose and lipids metabolism,inflammation response,pro liferation and differentiation.Som e d rugs targeted on PPARs,such as lipid-lowering and antidiabetic d rugs have been deve loped.Som e PPAR agonists were found carcinogenic in anim a l experim ents,including PPARαagonist fibrates,PPARγagonist thiazo lidinediones,PPARα/γdual agonist com pounds,and PPARδagonist compounds for c linical deve lopment.PPARαagonist carcinogenicity is associated w ith PPAR recep tor activation that regulates lipid m etabolism,and leads to lipids abnorm alities and increase by peroxisome oxidase in reactive oxygen species(ROS),causing DNA damage.Kup ffer cells can generate ROS by NADPH oxidase that promotes hepatocyte proliferation and inhibition of apoptosis.PPARγagonist carcinogenicity is generally caused by bladder stone.The carcinogenicity of PPAR agonists to humans has notbeen confirmed,but the carcinogenic potentialof these drugs cannot be ignored.
peroxisom e p ro liferators activated receptors agonists;toxic actions;carcinogens
WU Ying-liang,Tel:(024)23986278,E-mail:yingliang-1016@163.com
R977.15,R99
A
1000-3002(2014)03-0455-07
Foundation item:The project supported by Na tional Science and Technology Major Projects(2013ZX09302304)
2013-10-12 接受日期:2014-04-20)
(本文编辑:齐春会)
国家科技重大专项(2013ZX09302304)
邢立国(1974-),男,博士,主要从事药物致癌性评价,Te l:(024)62353435,E-mail:liguo-xing@163.com;吴英良(1957-),男,教授,博士生导师,主要从事神经精神药物药理学研究。
吴英良,Tel:(024)23986278,E-mail:yingliang-1016@163.com
猜你喜欢
黑龙江大学自然科学学报(2022年4期)2022-11-17 08:07:52
河北科技师范学院学报(2022年2期)2022-08-26 08:55:46
家庭医药(2022年9期)2022-05-30 10:48:04
食品安全导刊(2019年2期)2019-04-26 01:22:28
中成药(2018年10期)2018-10-26 03:41:22
天然产物研究与开发(2018年6期)2018-07-09 06:01:46
生命与灾害(2018年6期)2018-03-29 12:36:32
中国塑料(2016年3期)2016-06-15 20:30:00
华东理工大学学报(自然科学版)(2015年4期)2015-12-01 04:00:36
医学研究杂志(2015年5期)2015-06-10 06:43:26
