
全基因组扩增技术原理及研究进展
2014-03-22 01:53潘孝明梁兴国
生物技术通报 2014年12期
潘孝明 梁兴国
(中国海洋大学食品科学与工程学院,青岛 266003)
全基因组扩增技术原理及研究进展
潘孝明 梁兴国
(中国海洋大学食品科学与工程学院,青岛 266003)
DNA芯片及下一代测序等高通量技术的发展极大促进了分子生物学技术在各领域的广泛应用,但此时样品数量与质量往往是制约研究进行的重要因素,全基因组扩增技术(Whole genome amplification, WGA)则可有效解决这一问题,其可使极微量目标基因组DNA同时得到扩增从而提供满足高通量分析研究所需的起始材料。高效可靠的全基因组扩增技术使基于单细胞水平的大规模全基因组分析成为现实,且随着研究的不断深入,对全基因组扩增技术提出了更高的要求。对过去常用的以及近期发展的全基因组扩增技术进行了概述,阐明了各种方法的原理,并对各种方法的特点从原理上进行了分析总结,以期为全基因组扩增技术的应用提供一定的参考。
全基因组扩增 单细胞基因组测序 基因型 基因组变异 突变检测
现代分子生物学的不断发展除了为生物学领域带来了革命性的突破之外,也被广泛应用于其他相关领域,极大地推动了诸如食品、医学、环境等学科的研究,成为各学科前沿研究中必备的强大工具。分子生物学方法大多是以DNA作为最主要的研究对象,而很多情况下可供分析使用的DNA数量极少,如胚胎植入前遗传学诊断(Preimplantation genetic diagnosis,PGD)可有效避免植入胚胎存在某些疾病而具有显著的优生学意义,而这一技术的实现仅仅允许使用1-2个胚胎细胞进行分析检测[1];在法医鉴定中往往仅能获得痕量DNA样品等[2],此时下一代测序(Next generation sequencing,NGS)与DNA芯片技术等高通量分析方法往往受限于样品量太少而无法发挥其作用[3],全基因组扩增技术(Whole genome amplification,WGA)能够实现对整个基因组的扩增以提供大量可供分析样品,因而为此类研究提供了强有力的支持,并且高灵敏度WGA技术的不断进步也极大促进了单细胞全基因组测序技术的
发展,使很多针对单细胞的大规模研究分析成为了可能,在疾病早期诊断、微卫星序列分析、杂合子丢失(Loss of heterozygosity,LOH)、突变检测以及生物进化研究中具有重要的意义。近年来应用WGA技术的单细胞分析日臻成熟且取得了显著成果[4-8],本文将对目前已经广泛应用的以及新出现的WGA方法从原理、应用等方面进行介绍,并对各自方法的优点以及存在的问题进行概括性总结。
1 基于PCR技术的WGA方法
1.1 引物延伸预扩增法
在所有WGA方法中引物延伸预扩增法(Primerextension preamplification,PEP)是较早出现的一种,Zhang等[9]最早应用PEP方法实现了单个单倍体细胞基因组的全扩增。PEP法是一种基于PCR技术发展而来的WGA方法,主要原理是使用15个碱基长的随机引物(N15,理论上有415种组合)在37℃的低退火温度下进行较长时间的退火,然后缓慢升温至55oC进行长时间的引物延伸,如此反复多个循环(图1)。Dietmaier等[10]对PEP方法进行了一定的改进,包括对模板DNA的提取方法、反应循环条件的优化以及高保真聚合酶的使用等,并成功实现同时对单个肿瘤细胞的多个突变位点进行分析[10,11];Anchordoquy等[12]通过PEP法对口腔细胞基因组全扩增后进行基因型分析,对多达1 890例等位基因的分析结果表明PEP法可提供足量优质的模板满足高通量基因型分析的要求。但由于PEP法使用随机引物以及较为不严格的PCR循环参数,因此可能导致不均衡的扩增结果产生[13,14]。

图1 引物延伸预扩增法原理示意图[9]
1.2 简并寡核苷酸引物PCR
与PEP法类似,简并寡核苷酸引物PCR法(Degenerate oligonucleotide primed PCR,DOP-PCR)也是一种基于PCR技术的全扩增方法(图2),最早由Telenius等[15]提出,其原理是使用部分简并的引物进行PCR反应(引物中间部分含有6个随机碱基),在最初的几个循环过程中使用较低的温度(~25℃)进行退火以确保引物与模板的结合,并缓慢升温至引物延伸温度进行引物延伸,完成最初几个循环后使用相对较高的退火温度(~55℃)进行多循环常规PCR反应,对DOP-PCR结果影响较大的是因素是聚合酶及引物浓度,因此需要进行优化以获得最佳实验结果[15,16]。Cheung等[17]应用DOP-PCR对不足1 ng的人类基因组DNA实现了数百倍的扩增,并提供满足上百次微卫星序列基因型分析所需的模板;Van等[18]对传统DOP-PCR进行了一定改进,同时使用4条简并引物进行扩增,且每条引物中间都含有8个随机碱基,从而增加了引物的简并性,并将DOP-PCR产物使用Roche GS-FLX平台进行下一代测序(NGS)从而实现了对研究较少的大豆品种(Sinpaldalkong 2)的基因组结构分析,表明DOPPCR法在对未知样品分析中具有一定潜力。DOPPCR法虽然使用相对PEP法更为严格的PCR反应条件,但引物与模板间结合的不确定性以及引物间的相互作用仍可能导致较低的全扩增灵敏度及较高的错误率[19]。因此,较大程度上限制了该方法的应用,近年来应用该方法的报道相对较少。

图2 简并寡核苷酸引物PCR原理示意图[15]
1.3 连接介导PCR
广义上连接介导PCR(ligation-mediated PCR,LM-PCR)指的是一类有接头连接过程参与的PCR反应,相对于PEP与DOP-PCR,连接介导PCR最初并非设计用来进行基因组全扩增,其最早是由Pfeifer等应用于体内足迹研究(in vivo footprinting)、甲基化分析、DNA损伤鉴定等[20-23],在此基础上对其稍加改变即可进行基因组全扩增反应[4,24]。总体而言LM-PCR全扩增技术需要通过3个步骤来实现:(1)模板DNA的片段化处理:可通过物理剪切或限制性内切酶处理使基因组DNA断裂成若干适合PCR扩增的片段。使用物理剪切法对DNA进行处理时因无法预测片段末端结构,因此需要对其进行平末端化处理以满足其与接头进行连接的条件[25],如使用限制性内切酶对模板进行剪切,因各片段末端组成已知,因此大多数情况下无需对片段进行处理[26];(2)模板片段与接头连接:根据酶切片段类型设计可与之连接的接头,在DNA连接酶的作用下与模板片段两端连接,接头中含有一段外源通用引物序列,用作下一步PCR反应中的引物;(3)全扩增PCR反应:连接成功的模板片段经引物延伸后两端形成了通用引物结合位点,因此可以外源引入的通用引物进行PCR反应,包括接头在内的模板片段可同时得到扩增(图3)。

图3 连接介导PCR原理示意图
对基因组的片段化处理是LM-PCR全扩增技术的关键步骤之一,其对接下来的连接反应及PCR反应具有较大影响,使用物理剪切法可较好保证片段长度的均一性,但需要进行片段平末端化处理。Tanabe等[27]在传统LM-PCR的基础上发展了PRSG法(Adaptor-ligation PCR of randomly sheared genomic DNA)对基因组进行全扩增,其使用HydroShear仪将基因组DNA随机剪切成0.5-2 kb的片段进行接头连接后扩增,通过对307个微卫星序列进行基因分型以及287个基因座进行CGH分析的结果表明PRSG法可准确高效的实现全基因组扩增。酶法剪切无需昂贵的仪器因此可在绝大多数实验室进行,但使用限制性内切酶对基因组进行酶切时无法预测其片段长度,尤其是无法完全避免较长片段的产生,针对这一问题Liu等[28]采用3种内切酶(DpnII,HhaI,RsaI)对基因组进行酶切,通过对石蜡包埋的肿瘤组织的全扩增结果表明LM-PCR全扩增覆盖率可达到96%,进一步将产物应用于CGH以及PCR-SSCP分析(PCR-single-strand conformation polymorphism)验证了其扩增产物的准确性,说明LM-PCR全扩增方法在对保存状况较差样品的分析方面具有巨大的潜力。相对于其他基于PCR技术的全扩增方法(如上述的PEP与DOP-PCR),LM-PCR法使用较为严格的扩增条件,且使用的是一条通用引物,所有连接片段间与通用引物结合的能力相同,因此可在很大程度上保证所有片段得到相同程度的高灵敏度扩增。目前已有Sigma公司应用该技术开发的GenomePlex系列试剂盒产品[4,24,29]。LM-PCR方法的优点是扩增效率极高,缺点是步骤相对繁琐,对复杂DNA序列(如富含GC结构的模板)的扩增可能出现偏差,并且由于模板片段间具有相同的黏性末端(或平末端),因此在接头连接过程中这些片段间会不可避免的发生“无序自连”,导致重组得到的片段较长而扩增失败。针对这一“自连”问题,作者所在课题组使用IIS型限制性内切酶对基因组进行酶切产生具有随机碱基黏性末端的片段,配合使用具有随机碱基黏性末端的接头,可极大减少自连现象发生的几率。以BbvI内切酶为例(识别位点GCAGC(8/12)↓),其可产生具有4碱基随机碱基的黏性末端NNNN,因此可将自连概率减少44=256
倍,并通过qPCR等技术验证了这一改良方法极高的扩增效率,各目标片段可较为均衡的扩增数百万倍[30]。
2 恒温全基因组扩增反应
2.1 多重置换扩增反应
基于恒温核酸扩增技术的多重置换扩增法(Multiple displacement amplification,MDA)的出现相对PCR类全扩增方法较晚,其基本原理是使用一条硫代修饰的六核苷酸随机引物(N6)在恒温条件下与基因组随机退火,并在具有强链置换活性的phi29 DNA聚合酶的作用下发生链置换扩增反应,置换产生的单链序列又可与随机引物任意退火延伸,形成超分支扩增结构[31],由于phi29 DNA聚合酶具有较强的持续合成DNA的能力,因此可持续合成长达50-100 kb的产物(图4)。Dean等[32]最早应用MDA法进行全基因组扩增,相对于PCR类方法,MDA法显示出极高的扩增灵敏度(1-10基因组拷贝),且通过对8个染色体位点的扩增检测结果显示各目标间扩增偏差可控制在3倍以内,远远小于DOP-PCR等方法4-6个数量级的扩增偏差。此后,有大量成功应用MDA法进行WGA的报道,如Hellani等[33]将MDA法成功应用于PGD研究;Kumar等[34]应用MDA法实现了对难于培养的伯纳特氏立克次氏体基因组的全扩增等,均显示出其极高的WGA效率。但其对模板质量的要求相对较高,在非最佳实验条件下亦可导致非均衡扩增的产生[35]。由于MDA法是一种恒温核酸扩增方法,可能存在一定的非特异性扩增问题,即体系内不含有模板时也会产生大量产物,针对这一问题Marcy等[36]使用极微量的MDA体系(60 μL)对单细胞基因组DNA进行全扩增;Pan等[37]通过向体系中加入一种特殊的海藻糖进行MDA反应,均对非特异性扩增产生了一定的抑制效果。
MDA法是目前公认应用最广泛的WGA方法,操作简单且对实验仪器的要求极低,且使用非预变性的模板时也可取得良好的WGA效果,可进一步简化实验操作及避免污染的产生[32],可广泛应用于基因组测序、CGH、SNPs分析等相关领域,目前针对该方法开发的产品较为成熟,具有代表性的为Qiagen公司的Repli-g系列全扩增试剂盒[38-40]。
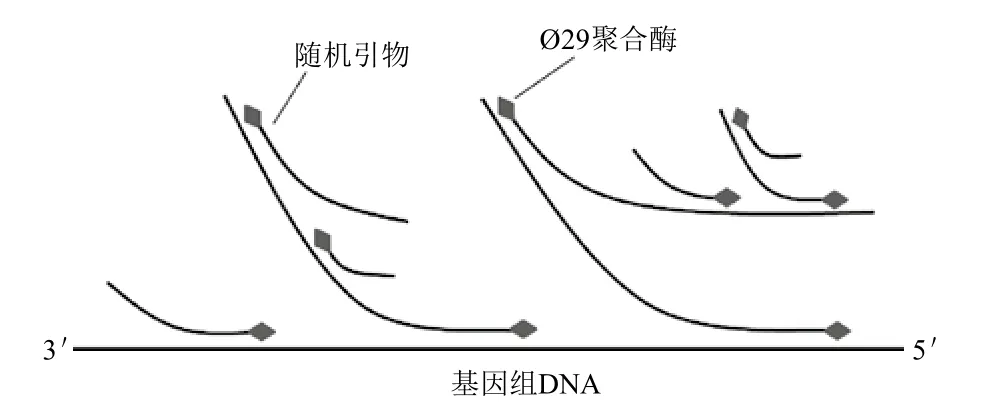
图4 多重链置换法全扩增原理示意图[31]
2.2 基于引物酶的全基因组扩增
由T7细菌噬菌体核酸复制方式演化而来的基于引物酶的全基因组扩增(Primer-based whole genome amplification,pWGA)是一种恒温全基因组扩增方法,相对于其他所有WGA方法,pWGA最大的特点是在扩增过程中不需要使用任何引物,且其不需要对模板进行预变性处理[41]。pWGA过程中所需的T7 gp4蛋白可同时发挥解旋酶和引物酶的作用,该蛋白的C端部分可利用脱氧胸苷三磷酸(dTTP)水解产生的能量来将DNA双链解旋;而N端则行使引物酶(primase)的作用,其特异性识别3'-CTGG(G/T)-5'和3'-CTGTG-5'并产生短的RNA作为扩增引物。引物生成后在T7 DNA聚合酶全酶的作用下进行持续的扩增反应,由T7 gp2.5基因编码的单链DNA结合蛋白(Single-stranded DNA binding protein)与解旋产生的单链DNA结合并与解旋酶/引物酶、聚合酶一起完成整个全基因组扩增过程(图5),该扩增模式与滚环扩增(Rolling circle amplification,RCA)相似并可以产生长达40 kb的产物[42]。
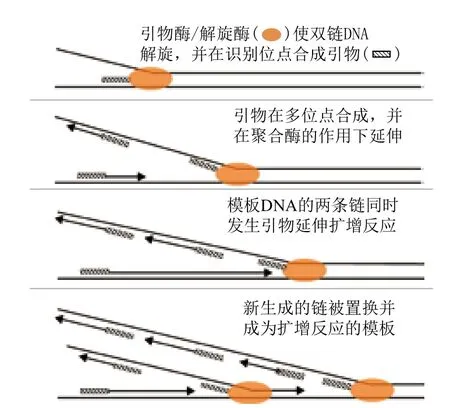
图5 基于引物酶全基因组扩增法原理示意图[42]
除上述几种蛋白(酶)外,pWGA反应还需要核苷二磷酸激酶(Nucleoside diphosphokinase)、无机焦磷酸酶(Inorganic pyrophosphatase)以及由肌酸激酶(Creatine kinase)和磷酸肌酸(Phosphocreatine)组成的ATP生成系统。其中核苷二磷酸激酶的主要作用是将NTPs中的γ-磷酸基团转移至NDPs以重新形成NTPs而平衡体系中的dNDP与dNTP的比例,并可以从一定程度上弥补解旋过程中造成的dTTP损失;无机焦磷酸酶可以将扩增反应中产生的无机焦磷酸分解为磷酸盐,减少因其累积而对T7 DNA聚合酶造成的抑制;最终肌酸激酶将磷酸肌酸中的高能磷酸键转移至dNTPs,从而合成新的dNTPs而提高全扩增的效率。
与MDA法相比,pWGA法仅需1 h即可完成反应。Schaerli等[43]发展了使用T4噬菌体gp61引物酶的全扩增体系T4 pWGA,并显示出在基因组测序、DNA标记、大规模菌落筛选以及DNA定量等方面的潜力,但相对于其他WGA方法,目前对pWGA法的报道仍偏少[2],除该方法面世不久之外,反应需要较多的蛋白(酶)、试剂组合也可能对其推广普及产生一定的限制作用。
3 多次退火环状循环扩增技术
美国国家科学院院士谢晓亮教授带领的团队于2012年底在Science杂志上连续发表了两篇应用其新发明的一种WGA方法进行单细胞测序研究的文章,引出了“多次退火环状循环扩增技术”(Multiple annealing and looping-based amplification cycles,MALBAC)这一全新的WGA方法,并应用该方法成功实现了单细胞水平的单核苷酸变异(Singlenucleotide variations,SNVs)以及拷贝数变异(Copynumber variations,CNVs)的研究[44,45]。其结合了MDA法与PCR方法的特点,使用的35 nt长的引物由一段固定的27 nt通用引物序列和8 nt随机碱基序列(N8)组成,在0oC时该8 nt随机碱基序列可与模板任意退火,梯度升温至65oC后在具有链置换活性的Bst大片段DNA聚合酶作用下发生链置换聚合反应,得到一系列长度不一的(0.5-1.5 kb)半扩增子(Semiamplicons),在94℃变性、0℃退火以及65℃延伸循环后,上一循环中半扩增子形成了两端具有互补结构(27 nt)的全扩增子(Amplicons),随后降低温度至58℃使得到的扩增子两端互补形成loop结构,从而可以避免引物与其进行结合导致全扩增子倍增导致的不均衡扩增,因此可以很大程度上保证该循环发生的是线性扩增。在经过5个上述类似线性扩增循环后可得到大量全扩增子并做为接下来PCR反应的模板,并使用该27 nt通用引物进行指数PCR反应,因此只有全扩增子才能得到有效扩增,从而实现对整个基因组的高效而又均衡的全扩增(图6)。

图6 多次退火环状循环扩增技术原理示意图[44]
MALBAC法显示出比MDA法更为均衡的扩增结果[44],但使用单细胞进行基因型分析时MALBAC法的假阳性率偏高,比MDA可高出40倍,这可能是因为MDA法使用的phi29 DNA聚合酶比MALBAC法应用的Bst和Taq聚合酶具有更高的保真度,因此往往需要使用多个细胞以获得更加准确的结果[46]。由于MALBAC法对单细胞全基因组扩增具有很高的覆盖率(93%)和均衡性,甫一出现就引起了较多的关注,Ni等[47]成功应用其对外周血液中极少数的循环肿瘤细胞(CTCs)进行CNVs分析,Hou等[48]对单个卵母细胞进行基因组测序,显示出该方法在癌症早期检测以及胚胎移植等领域的广阔应用前景。
4 结语
综上所述,单细胞基因组测序技术的发展极大促进了癌症、基因组变异等众多领域研究进展,而
决定这一技术应用成功与否的最关键因素就是全基因组扩增效果,因此对现有WGA方法进行改良或研发新的高效WGA方法是目前研究的热点[46,49-51]。总体而言现有WGA方法各有特点(表1),需根据实际情况选择合适的WGA方法加以应用。如PCR类全扩增方法对样品质量要求相对较低且扩增效率高,但全扩增产物片段较短,指数反应易导致所有片段的非均衡扩增;而MDA法的成功应用的前提是高质量基因组模板的获得,其扩增产物较为均衡且更加利于全基因组测序;但无论PCR类方法或是恒温全扩增方法,目前都存在一定的非特异性扩增现象,即使在较为严格的PCR反应条件下这一问题仍无法完全避免[52,53],因此对现有方法加以改进抑制非特异性扩增也是当前WGA方法研究中需要解决的问题,如数字微滴PCR(Digital microdroplet PCR)的发展可为基于PCR技术的WGA方法带来一些新的思路。

表1 主要的WGA方法比较
目前已有上千种生物基因组被测序,但是相对于更多的其他物种,已测序的物种数量仅占极少数[54],如自然界中绝大多数微生物不可培养且难以富集,导致针对这类微生物的全基因组测序难度极大[55,56]。因此,高效而又可靠的WGA技术将在更多相关研究领域中大有可为。
[1]Harper JC, Sengupta SB. Preimplantation genetic diagnosis:state of the art 2011[J]. Human Genetics, 2012, 131(2):175-186.
[2]蔡海强, 柳海涛, 史斌, 等. 全基因组扩增技术及其在法医个体识别中的应用[J].遗传, 2010, 32(11):1119-1125.
[3]潘星华, 朱海英, Marjani SL. 单细胞基因组学分析的技术前沿[J].遗传, 2011, 33(1):17-24.
[4]Lasken RS, Egholm M. Whole genome amplification:abundant supplies of DNA from precious samples or clinical specimens[J]. Trends in Biotechnology, 2003, 21(12):531-535.
[5]Hawkins TL, Detter JC, Richardson PM. Whole genome amplification-applications and advances[J]. Current Opinion in Biotechnology, 2002, 13(1):65-67.
[6]Hasmats J, Green H, Orear C, et al. Assessment of whole genome amplification for sequence capture and massively parallel sequencing[J]. PloS One, 2014, 9(1):e84785.
[7]Kumar S, Gangoliya SR, Berri M, et al. Whole genome amplification of the obligate intracellular pathogen coxiella burnetii using multiple displacement amplification[J]. Journal of Microbiological Methods, 2013, 95(2013):368-372.
[8]Maciejewska A, Jakubowska J, Pawlowski R. Whole genome amplification of degraded and nondegraded DNA for forensic purposes[J]. International Journal of Legal Medicine, 2013, 127(2):309-319.
[9]Zhang L, Cui X, Schmitt K, et al. Whole genome amplification from a single cell-implications for genetic analysis[J]. Proceedings of the National Academy of Sciences of the USA, 1992, 89:5847-5851.
[10]Dietmaier W, Hartmann A, Wallinger S, et al. Multiple mutation analyses in single tumor cells with improved whole genome amplification[J]. Am J Pathol, 1999, 154(1):83-95.
[11]Arneson N, Hughes S, Houlston R, et al. Whole-genome amplification by improved primer extension preamplification PCR(I-PEPPCR)[J]. CSH Protoc, 2008:pdb prot4921.
[12]Anchordoquy HC, McGeary C, Liu L, et al. Genotyping of three candidate genes after whole-genome preamplification of DNA collected from buccal cells[J]. Behavior Genetics, 2003, 33(1):73-78.
[13]Hanson EK, Ballantyne J. Whole genome amplification strategy for forensic genetic analysis using single or few cell equivalents of genomic DNA[J]. Analytical Biochemistry, 2005, 346(2):246-257.
[14]Pinard R, DeWinter A, Sarkis, GJ, et al. Assessment of whole genome amplification-induced bias through high-throughput, massively parallel whole genome sequencing[J]. BMC Genomics, 2006, 7:216.
[15]Telenius H, Carter NP, Bebb CE, et al. Degenerate oligonucleotideprimed PCR:general amplification of target DNA by a single degenerate primer[J]. Genomics, 1992, 13(3):718-725.
[16]Huang Q, Schantz SP, Rao PH, et al. Improving degenerate oligonucleotide primed PCR-comparative genomic hybridization for analysis of DNA copy number changes in tumors[J]. Genes Chromosomes & Cancer, 2000, 28(4):395-403.
[17]Cheung VG, Nelson SF. Whole genome amplification using a degenerate oligonucleotide primer allows hundreds of genotypes to be performed on less than one nanogram of genomic DNA[J]. Proc Nat Acad Sci USA, 1996, 93(25):14676-14679.
[18]Van K, Kang YJ, Shim SR, et al. Genome-wide scan of the soybean genome using degenerate oligonucleotide primed PCR:an example for studying large complex genome structure[J]. Genes & Genomics, 2012, 34(5):467-474.
[19]Lee CI, Leong SH, Png AE, et al. An isothermal method for whole genome amplification of fresh and degraded DNA for comparative genomic hybridization, genotyping and mutation detection[J]. DNA Research, 2006, 13(2):77-88.
[20]Pfeifer GP, Steigerwald SD, Mueller PR, et al. Genomic sequencing and methylation analysis by ligation mediated PCR[J]. Science, 1989, 246(4931):810-813.
[21]Dai SM, Chen HH, Chang C, et al. Ligation-mediated PCR for quantitative in vivo footprinting[J]. Nature Biotechnology, 2000, 18(10):1108-1111.
[22]Hornstra IK, Yang TP. In vivo footprinting and genomic sequencing by ligation-mediated PCR[J]. Analytical Biochemistry, 1993, 213(2):179-193.
[23]Quivy JP, Becker PB. An improved protocol for genomic sequencing and footprinting by ligation-mediated PCR[J]. Nucleic Acids Research, 1993, 21(11):2779-2781.
[24]Klein CA, Schmidt-Kittler O, Schardt JA, et al. Comparative genomic hybridization, loss of heterozygosity, and DNA sequence analysis of single cells[J]. Proc Natl Acad Sci USA, 1999, 96(8):4494-4499.
[25]Arneson N, Hughes S, Houlston R, et al. Whole-genome amplification by adaptor-ligation PCR of randomly sheared genomic DNA(PRSG)[J]. Cold Spring Harbor Protocols, 2008:pdb prot4922.
[26]Brugman MH, Suerth JD, Rothe M, et al. Evaluating a ligationmediated PCR and pyrosequencing method for the detection of clonal contribution in polyclonal retrovirally transduced samples[J]. Human Gene Therapy Methods, 2013, 24(2):68-79.
[27]Tanabe C, Aoyagi K, Sakiyama T, et al. Evaluation of a wholegenome amplification method based on adaptor-ligation PCR of randomly sheared genomic DNA[J]. Genes Chromosomes & Cancer, 2003, 38(2):168-176.
[28]Liu D, Liu C, DeVries S, et al. LM-PCR permits highly representative whole genome amplification of DNA isolated from small number of cells and paraffin-embedded tumor tissue sections[J]. Diagnostic Molecular Pathology, 2004, 13(2):105-115.
[29]Brueck C, Song S, Collins J. Oligonucleotide array CGH analysis of a robust whole genome amplification method[J]. Biotechniques, 2007, 42(2):230-233.
[30]Pan X, Wan B, Li C, et al. A novel whole genome amplification method using type IIS restriction enzymes to create overhangs with random sequences[J]. Journal of Biotechnology, 2014, 184:1-6.
[31]Hosono S, Faruqi AF, Dean FB, et al. Unbiased whole-genome amplification directly from clinical samples[J]. Genome Research, 2003, 13(5):954-964.
[32]Dean FB, Hosono S, Fang L, et al. Comprehensive human genome amplification using multiple displacement amplification[J]. Proceedings of the National Academy of Sciences of the USA, 2002, 99(8):5261-5266.
[33]Hellani AM, Akoum SM, Fadel ES, et al. Successful pregnancies after combined human leukocyte antigen direct genotyping and preimplantation genetic diagnosis utilizing multiple displacement amplification[J]. Saudi Medical Journal, 2012, 33(10):1059-1064.
[34]Kumar S, Gangoliya SR, Berri M, et al. Whole genome amplification of the obligate intracellular pathogen coxiella burnetii using multiple displacement amplification[J]. Journal of Microbiological Methods, 2013.
[35]Yilmaz S, Allgaier M, Hugenholtz P. Multiple displacement amplification compromises quantitative analysis of metagenomes[J]. Nature Methods, 2010, 7(12):943-944.
[36]Marcy Y, Ishoey T, Lasken RS, et al. Nanoliter reactors improve multiple displacement amplification of genomes from single cells[J]. PLoS Genetics, 2007, 3(9):1702-1708.
[37]Pan X, Urban AE, Palejev D, et al. A procedure for highly specific, sensitive, and unbiased whole-genome amplification[J]. Proceedings of the National Academy of Sciences of the USA, 2008, 105(40):15499-15504.
[38]Fernandez-Ortuno D, Tores JA, DeVicente A, et al. Multiple displacement amplification, a powerful tool for molecular genetic analysis of powdery mildew fungi[J]. Current Genetics, 2007, 51(3):209-219.
[39]Dickson PA, Montgomery GW, Henders A, et al. Evaluation of multiple displacement amplification in a 5 cm STR genome-wide scan[J]. Nucleic Acids Research, 2005, 33(13):e119.
[40]Ellegaard KM, Klasson L, Andersson SG. Testing the reproducibility of multiple displacement amplification on genomes of clonal endosymbiont populations[J]. PloS One, 2013, 8(11):e82319.
[41]Kim J, Easley CJ. Isothermal DNA amplification in bioanalysis:strategies and applications[J]. Bioanalysis, 2011, 3(2):227-239.
[42]Li Y, Kim HJ, Zheng C, et al. Primase-based whole genome amplification[J]. Nucleic Acids Research, 2008, 36(13):e79.
[43]Schaerli Y, Stein V, Spiering MM, et al. Isothermal DNA amplification using the T4 replisome:circular nicking endonuclease-dependent amplification and primase-based whole-genome amplification[J]. Nucleic Acids Research, 2010, 38(22):e201.
[44]Zong C, Lu S, Chapman AR, et al. Genome-wide detection of single-nucleotide and copy-number variations of a single human cell[J]. Science, 2012, 338(6114):1622-1626.
[45]Lu S, Zong C, Fan W, et al. Probing meiotic recombination and aneuploidy of single sperm cells by whole-genome sequencing[J]. Science, 2012, 338(6114):1627-1630.
[46]Lasken RS. Single-cell sequencing in its prime[J]. Nature Biotechnology, 2013, 31(3):211-212.
[47]Ni XH, Zhuo ML, Su Z, et al. Reproducible copy number variation patterns among single circulating tumor cells of lung cancer patients[J]. Proceedings of the National Academy of Sciences of the USA, 2013, 110(52):21083-21088.
[48]Hou Y, Fan W, Yan L, et al. Genome analyses of single human oocytes[J]. Cell, 2013, 155(7):1492-1506.
[49]Caldas C. Cancer sequencing unravels clonal evolution[J]. Nature Biotechnology, 2012, 30(5):408-410.
[50]Evrony GD, Cai X, Lee E, et al. Single-neuron sequencing analysis of L1 retrotransposition and somatic mutation in the human brain[J]. Cell, 2012, 151(3):483-496.
[51]Junker JP, van Oudenaarden A. Every cell is special:genomewide studies add a new dimension to single-cell biology[J]. Cell, 2014, 157(1):8-11.
[52]Kato T, Liang X, Asanuma H. Model of elongation of short DNA sequence by thermophilic DNA polymerase under isothermal conditions[J]. Biochemistry, 2012, 51(40):7846-7853.
[53]王阳, 贾蕾敏, 董平, 等. 三核苷酸双链重复序列扩展合成特性及其机理[J]. 生物化学与生物物理进展, 2013, 40(4):345-355.
[54]彭伶俐, 王琴, 辛明秀. 自然界中不可培养微生物的研究进展[J]. 微生物学杂志, 2011, 31(2):75-79.
[55]Zhang K, Martiny AC, Reppas NB, et al. Sequencing genomes from single cells by polymerase cloning[J]. Nature Biotechnology, 2006, 24(6):680-686.
[56]Kalisky T, Quake SR. Single-cell genomics[J]. Nature Methods, 2011, 8(4):311-314.
(责任编辑 狄艳红)
Principle of Whole Genome Amplification Technology and Its Progress
Pan Xiaoming Liang Xingguo
(College of Food Science and Engineering,Ocean University of China,Qingdao 266003)
The rapid developments of DNA array and next generation sequencing technologies had greatly promoted molecular biology techniques to be used in various fields. However, the limited quantity and quality of sample DNA may affect large scale genetic analyses. To overcome these problems, the whole genome amplification(WGA)technology was employed to amplify trace amounts of genomic DNA to sufficient quantity to meet the requirements of high throughput analyses. The highly efficient and reliable WGA methods also helped to realize large scale of genetic analyses based on even single cell. Moreover, as the single cell level research continues, more efficient WGA approaches were developed. This paper summarized the general WGA methods frequently used before and newly developed methods on basic principle level as well as the advantages and disadvantages of each method, which helped to make good use of the powerful WGA techniques.
Whole genome amplification Single cell sequencing Genotyping Genome variations Mutation detection
10.13560/j.cnki.biotech.bull.1985.2014.12.008
2014-04-10
国家青年千人计划,山东省万人计划,山东省自然科学杰出青年基金(JQ201204)
潘孝明,男,博士研究生,研究方向:食品分子生物学;E-mail:pan.xiao.ming@163.com
梁兴国,男,教授,研究方向:核酸化学与生物技术;E-mail:liangxg@ouc.edu.cn
猜你喜欢
中华骨与关节外科杂志(2022年1期)2022-08-31
今日农业(2021年11期)2021-08-13
科学(2020年4期)2020-11-26
教学考试(高考生物)(2020年6期)2020-11-23
中西医结合肝病杂志(2020年2期)2020-10-27
食品与生物技术学报(2020年8期)2020-01-06
科学24小时(2019年5期)2019-06-11
发明与创新(2019年9期)2019-03-26
中成药(2018年7期)2018-08-04
化学工业与工程(2015年1期)2015-02-10
