
Surgical menopause enhances hippocampal amyloidogenesis follow ing global cerebral ischem ia
2014-03-21 03:20ErinSottQuanGuangZhangYanDongDongHanRuiinWangRatnaVadlamudiDarrellBrann
Erin L.Sott,Quan-Guang Zhang,Yan Dong,Dong Han,Rui-M in Wang, Ratna K.Vadlamudi,Darrell W.Brann,*
aUniversity System of Georgia MD/PhD Program,Georgia Regents University,Augusta,GA 30912,USA
bInstitute of Molecular Medicine and Genetics,Georgia Regents University,Augusta,GA 30912,USA
cDepartment of Obstetrics and Gynecology,University of Texas Health Science Center at San Antonio,San Antonio,TX 78229,USA
Surgical menopause enhances hippocampal amyloidogenesis follow ing global cerebral ischem ia
Erin L.Scotta,b,Quan-Guang Zhangb,Yan Dongb,Dong Hanb,Rui-M in Wangb, Ratna K.Vadlamudic,Darrell W.Brannb,*
aUniversity System of Georgia MD/PhD Program,Georgia Regents University,Augusta,GA 30912,USA
bInstitute of Molecular Medicine and Genetics,Georgia Regents University,Augusta,GA 30912,USA
cDepartment of Obstetrics and Gynecology,University of Texas Health Science Center at San Antonio,San Antonio,TX 78229,USA
Background:Prematurely menopausal women have a doubled lifetime risk of dementia and a 5-fold increased risk of mortality from neurologicaldisorders,but the molecularmechanisms underlying these risks remain unclear.We hypothesized that ischem ia-induced amyloidogenesis may be enhanced in the hippocampus follow ing prolonged loss of ovarian 17β-estradiol(E2),which could contribute to these phenomena.
Methods:The currentstudy used a ratmodelof premature surgicalmenopause(10-week bilateralovariectomy)w ith E2 therapy either initiated immediately(short-term E2 deprivation(STED))or delayed to the end of the ovariectomy period(long-term E2 deprivation(LTED)).One week after continuous,subcutaneous E2 therapy,we subjected animals to 10-m in global cerebral ischem ia(GCI)to assess the effect of LTED on ischem ia-induced amyloidogenesis in the hippocampal CA1.
Results:The present study revealed that while hippocampalβ-amyloid(Aβ)is not typically enhanced follow ing GCI,there is a rapid,robust elevation of endogenous Aβin LTED females after GCI.In STED females,we observed that GCIattenuates and E2 maintains A Disintegrin and Metalloprotease 10(ADAM 10)expression in the hippocampal CA1,and concurrently,GCI increases and E2 decreases BACE1 levels in the same region.Intriguingly,however,we observed a loss of E2 regulation of ADAM 10,ADAM 17,and BACE1 levels in the hippocampalCA1 of LTED females,which provides mechanistic evidence for the enhanced post-ischem ic Aβload follow ing LTED.We also observed loss of E2 regulation of tau hyperphosphorylation in LTED females subjected to GCI.
Conclusion:Collectively,these studies partially explain the enhanced risk of dementia and mortality from neurological disorders seen in prematurely menopausal women and support timely initiation of E2 therapy to yield maximum neurological benefi t.
CopyrightⒸ2014,Shanghai University of Sport.Production and hosting by Elsevier B.V.A ll rights reserved.
Amyloid;Dementia;Estrogen;Ischem ia;Surgical menopause
1.Introduction
Women that enter menopause prematurely,or before the age of 40,due to bilateral oophorectomy incur a doubled lifetime risk of dementia and a 5-fold increased risk of mortality from neurological disorders.1,2The molecular mechanisms underlying the enhanced risks remain poorly understood,but prolonged loss of the neuroprotective ovarian steroid hormone 17β-estradiol(E2 or estrogen)is thought to play a key role,as estrogen therapy adm inistered at the time of surgery and continued until the median age of naturalmenopausal onset normalizes these risks.3Studies in our lab have provided a potential clue as to why surgical menopause may lead to an increased risk of dementia and mortality from neurological disorders.Along these lines,recent work has shown that the hippocampus sustains more damage from global cerebral ischemia(GCI)follow ing 10-week ovariectomy(long-term E2 deprivation(LTED));this includes previously unseen neuronal cell death in the hippocampal CA3 region,which is usually highly resistant to GCI,and a worse cognitive outcome follow ing GCI.4Intriguingly,additional work demonstrated that the hippocampus may become more susceptible to non-ischem ic stressors follow ing LTED as well, since the hippocampus of LTED female rats was also significantly more damaged follow ing exposure to β-amyloid (Aβ)(1—42),the most neurotoxic form of amyloid.4
Of the different types of dementia,Alzheimer’s disease (AD)is the most common,and it is characterized by two neuropathological hallmarks:senile plaques of Aβ and neurofibrillary tangles(NFTs)of hyperphosphorylated tau.5Excess neural deposits of Aβand NFTs are neurotoxic, causing extensive synapse loss and neurodegeneration,as well as an irreversible cascade of progressive memory loss,psychological disturbances,motor dysfunction,and eventually, death.6The amount of Aβpresent in the brain is largely dependent on the processing of amyloid precursor protein (APP),a Type I transmembrane protein that is sequentially cleaved by enzymes to create intracellular and extracellular fragments.7—9APP has two main processing pathways:nonamyloidogenic and amyloidogenic. During nonamyloidogenic processing,APP is sequentially cleaved w ithin the Aβsequence domain by anα-secretase,such as A Disintegrin and Metalloprotease 10 or 17(ADAM 10 or ADAM 17),followed by a gamma secretase enzyme complex.7,9As such,non-amyloidogenic APP processing precludes formation of Aβ and produces three non-toxic fragments.7,8Conversely,during amyloidogenic processing, APP is cleaved by theβ-secretase Aβcleaving enzyme 1 (BACE1)prior to cleavage by theγ-secretase machinery.This results in the formation of the insoluble,neurotoxic 40—42 am ino acid Aβprotein.If not successfully cleared from the brain,Aβmonomers form oligomers that then aggregate into extracellular deposits termed senile plaques.7
Intriguingly,E2 has been credited w ith a protective role in AD.10Observational studies revealed that postmenopausal women exposed to exogenous estrogens m id-life had a 29%—44%decreased risk of dementia,11—13and a recentstudy suggested that longer cumulative lifetime durations of estrogen exposure,including both endogenous and exogenous sources of E2,were associated w ith a lowered risk of AD,w ith each additional month of E2 exposure translating to a 0.5% decrease in AD risk.14W ith respect to basic science studies, E2 has also been repeatedly shown to protect against the neuropathological hallmarks of AD bothinvitroandin vivo.10,15For instance,E2 was found to prevent phosphorylation of the m icrotubule-associated protein tau follow ing cerebral ischem ia in rodents,which m itigates subsequent formation of NFTs.15,16Furthermore,exogenous E2 is well known to protect against Aβneurotoxicity,15,17,18and brainspecific E2 depletion was found to accelerate Aβdeposition and hinder Aβclearance in a transgenic mouse model of AD.19Collectively,these studies suggest that E2 tends to reduce the neural load of Aβ,and they corroborate postmortem studies,which found significantly reduced levels of E2 in the brains of female AD patients,compared w ith ageand gender-matched controls.19In regard to the mechanism(s) through which E2 modulates neural Aβ,scientific evidence supports E2 influence ofboth Aβdeposition and Aβclearance. Along these lines,E2 is purported to regulate expression of at least two major proteins responsible for removalof neurotoxic Aβ:insulin degrading enzyme and neprilysin.20—24W ith respect to Aβdeposition,several studies suggest that E2 may regulate APP processing at several steps,thereby promoting the non-amyloidogenic pathway.As evidence,BACE1,the rate-lim iting enzyme for Aβformation,has several estrogen response elements(EREs)w ithin its promoter region,25and E2 has been shown to decrease BACE1 expression both in mixed neuronal cultures and in neuronsin vivo.15,20,26,27Conversely,E2 has also been hypothesized to regulate two putativeα-secretases ADAM 104,27-30and ADAM 17,26,31which is also known as TNFα-converting enzyme(TACE).
While E2’s neuroprotective role in AD has been well studiedin vitro,E2’s neuroprotection from AD has not been completely characterizedin vivo,particularly considering the development of AD-like neuropathology follow ing GCI. Furthermore,aside from a single observed decrease of neprilysin expression in the brain 45 days post-ovariectomy,24and our lab’s recent finding of a sw itch to amyloidogenic APP processing in the hippocampal CA3 region follow ing GCI in long-term ovariectom ized females,4the effect of LTED(surgical menopause)on critical pathways affecting Aβload in non-transgenic rodents is largely unknown.A long these lines, the current study attempted to determ ine whether surgical menopause enhanced amyloidogenesis in the hippocampal CA1 following a stressor(GCI).Furthermore,the current study also aimed to definitively characterize acute E2 regulation of APP processing(ADAM 10,ADAM 17,and BACE1 expression)in the hippocampal CA1 follow ing GCI and to determ ine whether E2 regulation of APP processing is lost follow ing long-term ovariectomy,as these events could mechanistically explain the enhanced risk of dementia and mortality from neurological disorders observed in prematurely menopausal women.
2.M ethods
2.1.Animals
A ll procedures were approved by the Georgia Regents University Institutional Animal Care and Use Comm ittee (Animal Use Protocols:09-03-174 and 2012-0474)and were conducted in accordance w ith the National Institutes of Health guidelines for animal research.Young adult(3-month-old) female Sprague—Daw ley rats were utilized for these studies. A ll animals were group housed on a 10 h/14 h light—darkcycle and fedad libitumusing Harlan’s 8604 Teklad Rodent Diet.
2.2.Surgicalmenopause,estrogen therapy,and GCI
To induce surgicalmenopause,all female rats were bilaterally ovariectom ized under isoflurane anesthesia.At the time of menopausal onset(short-term E2 deprivation(STED))or 10 weeks later(LTED)the animals had subcutaneous,osmotic m ini-pumps(0.5μL/h,14-day release;A lzet,CA,USA) implanted between their scapulae containing either placebo (20%β-cyclodextrin)or17β-estradiol(0.0167 mg E2 in 20%βcyclodextrin)to m im ic physiologicalE2 levels produced during Diestrus I(10—15 pg/m L serum).16As such,STED rats modeled surgical menopause w ith immediate E2 therapy and LTED modeled surgical menopause w ith delayed E2 therapy. Oneweek aftercontinuoustreatment,GCIwasperformed using 4-vesselocclusion asdescribed previously.32—34Briefl y,the day before GCI,animals were anesthetized using intraperitoneal chloral hydrate(350 mg/kg)or intraperitoneal ketam ine/xylazine(60 mg/kg and 8 mg/kg,respectively),and both vertebral arteries(VA)were permanently occluded atthe levelof the alar foram ina via electrocauterization.Immediately follow ing bilateral VA occlusion,both common carotid arteries(CCA) were carefully isolated and loosely ligated w ith suture thread w ithout interrupting blood flow.A fter a 24-h recovery period, animals were re-anesthetized with light isoflurane(1%—4%), and the bilateral CCA were occluded with hemostatic clips to induce 10 m in of complete forebrain ischem ia.Animals which lost their righting reflex w ithin 30 s and whose pupils were dilated and unresponsive to lightduring cerebralischem ia were selected for the experiments.After 10 m in,the clips were removed,and reperfusion was confi rmed before the wound was sutured.During GCI,rectal temperature was maintained at 36.5°C—37.5°C w ith a thermalblanket.Sham animals did not receivepumpsand underwentidenticalGCIsurgicalprocedures except that the CCAwere simply exposed butnotoccluded.
2.3.Histological sample preparation
For brain harvesting,animals were deeply anesthetized w ith isoflurane and transcardially perfused w ith 0.9%saline at 3 h or 24 h post ischemia-reperfusion,followed by fixation w ith cold 4%paraformaldehyde in 0.1 mol/L phosphate buffer (PB).Brains were post-fixed in the same fixative overnightat 4°C and cryoprotected w ith 30%sucrose in 0.1 mol/L PB,pH 7.4 for 24—36 h.Coronal sections(25μm)were collected throughout the entire dorsal hippocampus(~2.5—4.5 mm posterior from Bregma,~100 sections per brain)for each animal.
2.4.Diaminobenzidine(DAB)staining
For DAB staining,sections were incubated w ith 10% normal horse serum in phosphate-buffered saline(PBS)containing 0.1%Triton X-100 and 0.3%H2O2for 1 h at room temperature to block nonspecific surfaces.Sections were then incubated w ith a single primary antibody:anti-PHF1(1:1000, gift from Peter Davies)or anti-Aβ(1:100,MAB8768;M illipore,Billerica,MA,USA)overnight at 4°C in PBS containing 0.1%Triton-X100 and 1%horse serum.Afterward, sections were washed w ith PBS containing 0.1%Triton-X100, followed by incubation w ith secondary biotinylated horse antirabbit/anti-mouse antibodies(1:500,Vector Laboratories,Inc., Burlingame,CA,USA)in the same buffer for 1 h at room temperature.Sections were then washed and incubated w ith ABC reagent for20 m in at room temperature.Finally,sections were rinsed,mounted onto a slide,and incubated w ith DAB reagent for 2—10 m in,according to the manufacturer’s instructions(Vector Laboratories,Inc.).Follow ing DAB incubation,slides were washed briefl y w ith distilled water, dehydrated in increasing concentrations of ethanol,cleared in xylene,and mounted using a xylene-based mounting medium. Images were captured on an Axiophot-2 visible/fluorescence m icroscope using an AxioVision 4Ac software system(Carl Zeiss,Jena,Germany).Analysis was performed by counting the number of immunopositive neurons per 250μm length of the medial CA1 pyramidal cell layer.A mean±SE was calculated foreach treatmentgroup,which consisted of four to seven animals each and three to five sections per animal.
2.5.Immunofluorescence staining
Coronal sections were incubated w ith 10%normal donkey serum for 1 h at room temperature in PBS containing 0.1% Triton X-100,followed by incubation w ith primary antibody: anti-ADAM 10(1:50,sc-25578;Santa Cruz Biotechnology, Inc.,Santa Cruz,CA,USA),anti-ADAM 17/TACE(1:50,sc-6416;Santa Cruz Biotechnology,Inc.),or anti-BACE1(1:100, AHB0241;Invitrogen,Carlsbad,CA,USA),overnightat4°C in the same buffer.After primary antibody incubation,sections were washed for 3×10 m in at room temperature,followed by incubation w ith the appropriate combination of secondary antibodies:Alexa-Fluor 488/568/647 donkey anti-rabbit/antimouse/anti-goat(1:500;Invitrogen).Sections were then washed w ith PBS containing 0.1%Triton X-100 for3×10 min, followed by 2×5 m in w ith 1×PBS and briefl y w ith water. Then,sections were mounted w ith water-based mounting medium containing anti-fading agents.
2.6.Confocal microscopy
A ll images were captured on an LSM 510 Meta confocal laser m icroscope(Carl Zeiss)using a 40× oil immersion Neofluor objective(NA,1.3)w ith the image size set at 1024×1024 pixels.The follow ing excitation lasers/em ission fi lters settings were used for various chromophores:argon/2 laser was used for A lexa-Fluor 488,w ith excitation maximum at 490 nm and em ission in the range of 505—530 nm,HeNe1 laser was used for A lexa-Fluor 568 w ith excitation maximum at 543 nm and em ission in the range of 568—615 nm,and HeNe2 laser was used for Alexa-Fluor 647 w ith excitation maximum at 633 nm and em ission in the range of 650—800 nm.The captured images were viewed and analyzedusing LSM 510 Meta imaging software.Simultaneous exam ination of negative controls confi rmed the absence of nonspecific immunofluorescent staining,cross-immunostaining,or fluorescence bleed-through.Images were analyzed by measuring the integrated density of fluorescent staining w ith ImageJ analysis software(Version 1.45s;http://imagej.nih. gov/ij/download.htm l,NIH,Bethesda,MD,USA)for each animal(2—5 sections/animal),and a mean±SE was calculated from the data in each group(n=5—10 animals/group).
2.7.Brain homogenization and protein extraction
For total protein extraction from the hippocampal CA1 region,rats were euthanized by rapid decapitation under deep anesthesia 3 h or 24 h post ischem ia-reperfusion.Once the whole brains were removed,the hippocampi were dissected from both sides of the hippocampal fissure,and the dorsal CA1 regions were separated.CA1 hippocampal tissues were immediately frozen in dry ice,and stored at-80°C untiluse. Tissues were homogenized w ith a Teflon-glass homogenizer in ice cold homogenization medium consisting of 50 mmol/L HEPES(pH 7.4),150 mmol/L NaCl,12 mmol/Lβ-glycerophosphate,3 mmol/L dithiothreitol(DTT),2 mmol/L sodium orthovanadate(Na3VO4),1 mmol/L EGTA,1 mmol/L NaF,1 mmol/L phenylmethylsulfonyl fl uoride(PMSF),1% Triton X-100,and inhibitors of proteases and enzymes (0.5 mmol/L PMSF,10μg/m L each of aprotinin,leupeptin, and pepstatin A).The homogenates were centrifuged at 15,000gfor 30 m in at 4°C,and supernatants were collected and stored at-80°C until use.Protein concentrations were determ ined w ith a Modified Low ry Protein Assay Kit (Thermo Scientific,Waltham,MA,USA),using bovine serum album in as a standard.
2.8.Western blotting
For Western blotting,20—50μg of total hippocampal CA1 protein lysate were separated via 4%—20%SDS-PAGE.Proteins were transferred to a polyvinylidene fluoride(PVDF) membrane(Immobilon-P;M illipore),blocked for 3 h,and incubated w ith 1°antibody against AβOligomers(1:500, AB9234;M illipore),PHF-1(1:1000,gift from Peter Davies), Tau(1:200,sc-1995;Santa Cruz Biotechnology),or Amyloid Precursor Protein C-Term inal Fragments(1:4000,A8717; Sigma—Aldrich,St.Louis,MO,USA)overnight at 4°C.α-Tubulin(1:500,sc-5286,Santa Cruz Biotechnology)served as a loading control.The membrane was then washed w ith Tween 20-PBS to remove unbound antibody and incubated w ith 2°antibody:Alexa Fluor 680/800 goat anti-rabbit/mouse IgG (1:10,000;Invitrogen)or Alexa Fluor 680/800 donkey antigoat IgG(1:10,000,Invitrogen),for 1 h at room temperature.Bound proteins were visualized using the Odyssey Imaging System(LI-COR Bioscience,Lincoln,NE,USA),and semi-quantitative analysis of the bands was performed using ImageJ analysis software.To quantitate hippocampal protein abundance,band densities of the indicated total proteins were analyzed and expressed as ratios relative to either full-length protein or α-tubulin signals, as appropriate, and a mean± SE was calculated from each group for graphical presentation and statistical comparison.
2.9.Statistical analysis
Statistical analysis was performed using two-way analysis of variance(ANOVA),followed by a Student-Newman-Keulspost-hoctest via NCSS software(NCSS,LLC.,www.ncss. com).Statistical significance was accepted at the 95%confidence level(p< 0.05).All data were expressed as mean±SE.
3.Results
3.1.Surgical menopause enhances development of AD-related neuropathology in the hippocampal CA1 region following GCI
We fi rstaimed to determ ine whether premature and chronic loss of ovarian E2 would enhance the developmentof AD-like neuropathology in the hippocampus following an ischem ic insult.To investigate,we utilized the STED and LTED animal models,w ith immediate(STED)ordelayed(LTED)E2 therapy, and used GCI as the stressor.We fi rst sought to determ ine whether the hippocampal load of Aβwas altered in LTED females subjected to GCI.DAB staining was used to visualize endogenous neuronal Aβin the STED and LTED hippocampal CA1 region ofnon-ischem ic sham and ischem ic(GCI)Pla-and E2-treated animals.The results revealed a robust increase in number of pyram idalcells immunopositive for intracellular Aβ oligomers 24 h post GCI in the hippocampal CA1 region of LTED,butnot STED,females(Fig.1A:e,f and B).Furthermore,Western blotting analysis also revealed significantly increased Aβoligomer formation in the hippocampal CA1 region of LTED female rats 24 h post GCI,relative toα-tubulin expression(Fig.1B),and this increase was not attenuated by delayed E2 treatmentin LTED females.These findings suggest that neuronal Aβload is increased in long-term surgically menopausalratssubjected to cerebralischemia and thatdelayed E2 therapy cannotprevent this event.
Since neurofibrillary tangles are another major neuropathological hallmark of AD,we next chose to exam ine E2’s ability to regulate the hyperphosphorylation of tau follow ing chronic loss of ovarian E2.Cerebral ischem ia is a well-known tauopathy.35—38In fact,we previously demonstrated that GCI induces significanthyperphosphorylation of tau 24 h post GCI and that low-dose E2 pretreatment attenuates this event.16We, thus,hypothesized that E2’s regulation of tau hyperphosphorylation may be lost follow ing LTED.To investigate, we exam ined paired helical fi laments(PHF)of m icrotubuleassociated tau phosphorylated at Ser 396 and Ser 404,two residues implicated in human AD neuropathology.Results revealed that both the number of PHF-immunopositive cells (Fig.2A)and PHF protein levels(Fig.2B)were increased 24 h after GCI(Fig.2A:b,e and B),and 1 week of E2 pretreatment,initiated immediately follow ing ovariectomy,wasable to prevent this event in STED rats(Fig.2A:c and B).In contrast,delayed E2 treatment was unable to m itigate the phosphorylation of tau at these two pathological residues in LTED rats(Fig.2A:f and B),suggesting that E2 regulation of tau phosphorylation is,indeed,lost follow ing LTED.
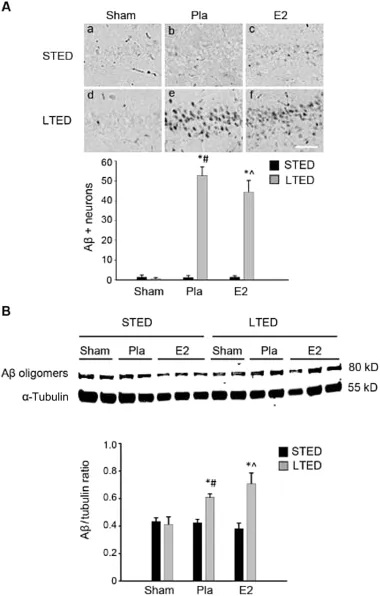
Fig.1.Post-ischem icβ-amyloid(Aβ)load is enhanced in the hippocampus follow ing long-term ovariectomy.(A)Representative DAB staining(top) demonstrates Aβexpression in the CA1 region 24 h post 10-m in global cerebral ischem ia.Quantitative summary(bottom,means±SE,n=4—7 animals/group)indicates the number of Aβ-positive neurons per 250μm medial CA1.(B)Western blotting shows Aβexpression 24 h post 10-m in global cerebral ischem ia in total CA1 hippocampal lysates of female rats treated with placebo(Pla)or low-dose estrogen(E2)either immediately(STED)or 10 weeks(LTED)follow ing bilateral ovariectomy.Quantifications of band densities(means±SE,n=4—7 animals/group)are expressed as densitometric ratios of Aβtoα-tubulin.*p<0.05,compared to sham;#p<0.05,compared to STED Pla;^p<0.05,compared to STED E2.Scale bar=50μm.
3.2.Surgical menopause leads to loss ofα-secretase expression in the hippocampal CA1 after GCI
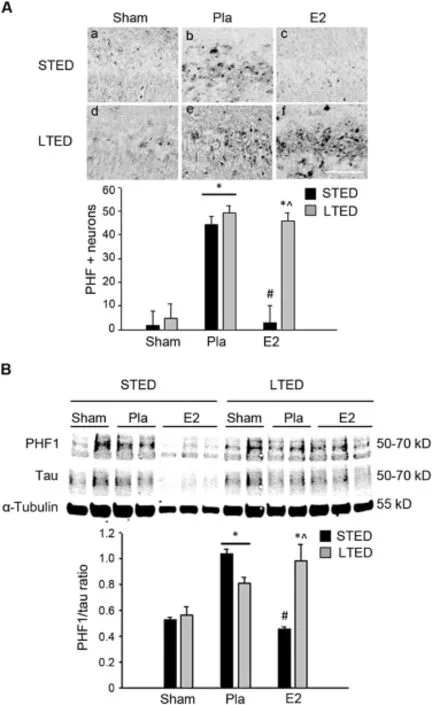
Fig.2.Estrogen regulation of ischem ia-induced tau phosphorylation is lost follow ing long-term ovariectomy.(A)Representative DAB staining(top) demonstrates expression of m icrotubule-associated protein tau phosphorylated atSer396 and Ser404(paired helical fi laments,PHF)in the hippocampalCA1 region 24 h after 10-m in global cerebral ischem ia.Quantitative summary (bottom,means±SE,n=4—7 animals/group)indicates the number of PHF-positive neurons per250μm medialCA1.(B)Western blotting shows PHF and total tau expression 24 h post 10-min global cerebral ischemia in total CA1 hippocampal lysates of female rats treated w ith placebo(Pla)or low-dose estrogen(E2)either immediately(STED)or 10 weeks(LTED)follow ing bilateralovariectomy.Tubulin is shown as a loading control.Quantifi cations of band densities(means± SE,n=4—7 animals/group)are expressed as densitometric ratios of PHF to tau.*p<0.05,compared to sham;#p<0.05, compared to STED Pla;^p<0.05,compared to STED E2.Scale bar=50μm.
To better understand the mechanisms underlying the marked elevation of endogenous Aβin LTED rats,we next exam ined hippocampal CA1 expression of two putativeαsecretases:ADAM 10 and ADAM 17,as well as theβ-secretase BACE1(section 3.3).ADAM 10 and ADAM 17 are thought to be the driving forces of non-amyloidogenic processing of APP,and although some controversy exists regarding which putativeα-secretase is mainly responsible,9recent studies have provided evidence that ADAM 10 is the primaryα-secretase and that ADAM 17 plays a more secondary role in the non-amyloidogenic processing of APP.39—41In our studies,we searched for evidence of acute E2 regulationof ADAM 10 and 17 expression follow ing GCI and/or dysregulation of ADAM 10 and ADAM 17 expression follow ing LTED.As shown in Fig.3,confocalm icroscopy revealed that ADAM 10 expression was significantly down-regulated in the hippocampal CA1 region 3 h post GCI(Fig.3A:b,e and B) and that immediate E2 replacement follow ing ovariectomy prevented this loss in STED females(Fig.3A:c and B).LTED placebo(Pla)animals had markedly decreased ADAM 10 levels 3 h following GCI,sim ilar to STED Pla animals.Of significant interest,however,delayed E2 therapy in LTED animals could notprevent the post-ischem ic loss of ADAM 10 in the hippocampal CA1(Fig.3A:f and B).
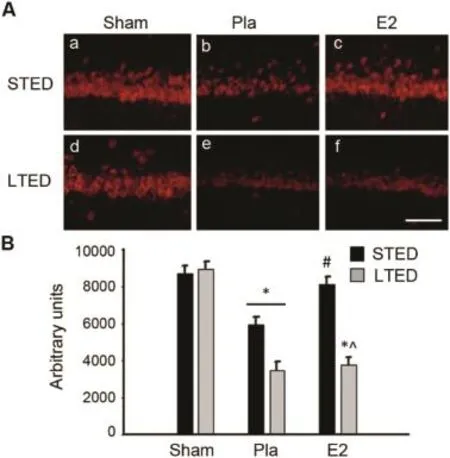
Fig.3.Hippocampal ADAM 10 expression is acutely regulated by global cerebral ischemia and estrogen butdysregulated after long-term ovariectomy. (A)Representative photom icrographs depict ADAM 10 expression in the hippocampal CA1 of short-term E2-deprived(STED)and long-term E2-deprived(LTED)female rats 3 h follow ing 10-m in global cerebral ischem ia. (B)Quantitative summary of data(means±SE,n=4—6 animals/group) indicates the raw integrated density of ADAM 10 immunostaining in the medial CA1.*p<0.05,compared to sham;#p<0.05,compared to STED placebo(Pla);^p<0.05,compared to STED E2.Scale bar=50μm.
While recent literature touts ADAM 10 as the mainαsecretase responsible for the non-amyloidogenic processing of APP,39,40other studies maintain that ADAM 17 or TACE plays a major role in the same process.42Therefore,we also exam ined expression of ADAM 17 follow ing GCI and longterm ovariectomy.In contrast to ADAM 10,there was no ischem ia-induced decrease of ADAM 17 expression in the hippocampus 3 h post GCI in STED females(Fig.4A:b and B).Furthermore,there was no E2 regulation of ADAM 17 expression in the hippocampal CA1 region at the same time point after ischem ia(Fig.4A:c and B).Follow ing 10-week ovariectomy,non-ischem ic LTED sham animals displayed a pattern for increased basal ADAM 17 immunostaining,but this trend did not reach statistical significance.However, confocal m icroscopy did reveal a marked loss of ADAM 17 expression 3 h post GCI in long-term ovariectom ized (LTED)females,and importantly,delayed E2 therapy was unable to prevent this loss(Fig.4A:e,f and B).As a whole, our data suggest that non-amyloidogenic processing of APP may be significantly impaired in the event of ischem ia follow ing long-term ovariectomy,as hippocampal CA1 region expression of bothα-secretases ADAM 10 and ADAM 17 are significantly decreased upon exposure to ischem ic stress in long-term ovariectom ized rats.Furthermore,the observed impairmentof non-amyloidogenic processing of APP suggests that a sw itch to amyloidogenic processing of APP may occur in LTED females in the event of ischem ic stress.
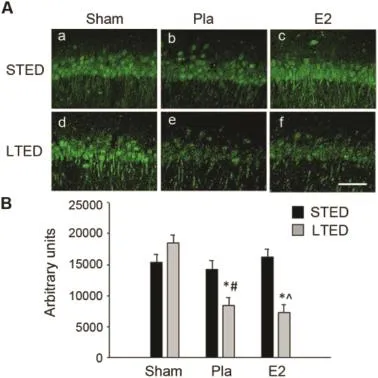
Fig.4.Hippocampal ADAM 17 expression is diminished after long-term ovariectomy in the event of global cerebral ischem ia.(A)Representative photomicrographs depict ADAM 17 expression in the hippocampal CA1 region 3 h after 10-m in global cerebral ischem ia.Note that a marked reduction in ADAM 17 expression was only observed in ischemic long-term ovariectom ized(LTED)females.(B)Quantitative summary of data(means±SE,n=4—6 animals/group)indicates the raw integrated density of ADAM 17 immunostaining in the medial CA1.*p<0.05,compared to sham;#p<0.05, compared to short-term E2-deprived(STED)placebo(Pla);^p< 0.05, compared to STED E2.Scale bar=50μm.
3.3.Surgical menopause enhances BACE1 expression and promotes a switch to amyloidogenic processing of APP in the hippocampal CA1 after GCI
Since theβ-secretase BACE1 is thought to be the ratelim iting step for Aβformation via the amyloidogenic processing of APP,25we next aimed to determ ine how GCI, exogenous E2,and long-term ovariectomy influence BACE1 expression in the hippocampal CA1 region.Confocal microscopy analysis revealed thatneuronal BACE1 expression was acutely up-regulated in the hippocampal CA1 region of STEDfemales 3 h follow ing GCI and that this elevation was prevented by pretreatment w ith low-dose E2 initiated at the time of ovariectomy(Fig.5A:a—c and B).However,follow ing LTED,we observed a loss of E2 regulation of ischemiainduced BACE1 expression in the hippocampal CA1 region (Fig.5A:e,f and B),suggesting that low-dose E2 is no longer able to prevent the ischem ia-induced increase of hippocampal BACE1 follow ing prolonged loss of ovarian E2.Furthermore, we noted a modest increase in basal expression of BACE1 in the hippocampal CA1 region of non-ischem ic LTED sham animals,but sim ilar to changes observed in basal ADAM 17 expression,this trend did not reach statistical significance (Fig.5A:d and B).These results agree w ith the aforementionedα-secretase results,suggesting that non-amyloidogenic processing of APP is significantly impaired and that amyloidogenic processing of APP is significantly enhanced follow ing long-term ovariectomy(LTED),particularly in the event of GCI.
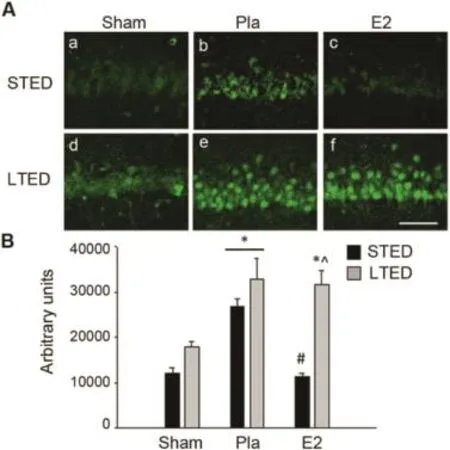
Fig.5.Long-term ovariectomy enhances BACE1 expression in the hippocampal CA1.(A)Representative photomicrographs demonstrate BACE1 expression in the hippocampal CA1 3 h post10-m in global cerebral ischem ia. Note the elevated expression of BACE1 in long-term ovariectom ized(LTED) females.(B)Quantitative summary of data(means±SE,n=4—6 animals/ group)indicates the raw integrated density of BACE1 immunostaining in the medialCA1.*p<0.05,compared to sham;#p<0.05,compared to short-term E2-deprived(STED)placebo(Pla);^p<0.05,compared to STED E2.Scale bar=50μm.
In light of the evidence suggesting a post-ischem ic sw itch to amyloidogenic APP processing follow ing surgical menopause,we next decided to more closely examine the proteolytic processing of APP.As discussed previously,APP processing can be categorized as either non-amyloidogenic or amyloidogenic.Non-amyloidogenic processing of APP occurs through sequential cleavage by α-and γ-secretases and produces three non-toxic fragments:p3,sAPPα,and C83.7In contrast,amyloidogenic APP processing occurs through sequential cleavage byβ-andγ-secretases and produces the neurotoxic Aβprotein,as wellas two other fragments:sAPPβ and C99.7To investigate changes in APP processing in the current study,we performed Western blotting analysis for the two APP C-Term inal fragments C99 and C83,which are representative of amyloidogenic and non-amyloidogenic APP processing,respectively,and we compared the C99/C83 ratio in the hippocampal CA1 region among the different treatment groups.As expected,the C99/C83 ratio was less than 1 in STED sham animals,suggesting thatnon-amyloidogenic APP processing predom inates in the hippocampus under basal conditions(Fig.6).This ratio was modestly elevated(1.0)24 h follow ing GCIin STED animals and returned to baseline if E2 therapy was adm inistered immediately follow ing ovariectomy (Fig.6),indicating that GCI promotes amyloidogenic and E2 promotes non-amyloidogenic processing of APP.While the C99/C83 ratio remained less than 1 in LTED sham animals, this ratio was significantly elevated(>1)in both LTED placebo-and LTED E2-treated females(Fig.6).This observation corroborates ourα-andβ-secretase data,suggesting that follow ing surgicalmenopause,GCIinduces a major switch to amyloidogenic APP processing in the hippocampal CA1 and that delayed E2 therapy is unable to m itigate this event.
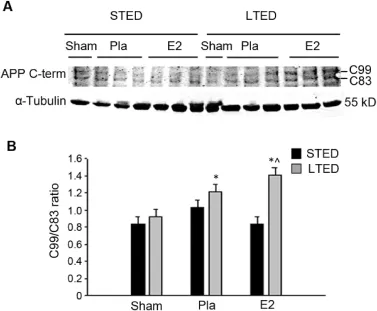
Fig.6.Long-term ovariectomy promotes a switch to amyloidogenic APP processing in the hippocampal CA1 follow ing global cerebral ischem ia.(A) Western blotting demonstrates C-Term inal fragments of amyloid precursor protein(APP)3 h following 10-m in global cerebral ischem ia in total CA1 hippocampal lysates of female rats treated w ith placebo(Pla)or low-dose estrogen(E2)either immediately(STED)or 10 weeks(LTED)follow ing bilateral ovariectomy.C99 is a byproduct ofβ-secretase-mediated APP cleavage,and C83 is a byproduct ofα-secretase-mediated APP cleavage. α-Tubulin is shown as a loading control.(B)Quantifications of band densities (means±SE,n=3—5 animals/group)are expressed as densitometric ratios of C99 to C83.Note that C99/C83 ratios greater than 1 were only observed in LTED females subjected to ischemia.*p<0.05,compared to sham;^p<0.05, compared to STED E2.
4.Discussion
The purpose of the current study was to test the hypothesis thatsurgicalmenopause leads to enhanced amyloidogenesis in the hippocampal CA1 region after ischem ic injury and to decreased sensitivity of the APP processing pathway to E2 regulation.Our study yielded several new findings,including: 1)acute regulation of theα-secretase ADAM 10 in the hippocampal CA1 region by both GCIand E2,2)evidence of an ischem ia-induced sw itch to amyloidogenic processing of APP in the hippocampal CA1 region,3)a robust increase in hippocampal Aβload in LTED female rats after an ischem ic insult,and 4)a loss of E2’s ability to regulate post-ischem ic changes in ADAM 10,ADAM 17,BACE1,and PHF follow ing LTED.Collectively,these findings suggest that E2’s ability to prevent post-ischemic hippocampal AD-related protein induction is,indeed,lost follow ing long-term ovariectomy.
W ith respect to the fi rst finding(acute regulation of ADAM 10 by GCI or E2),expression of theα-secretase ADAM 10 has been shown to be decreased by oxygenglucose-deprivation,chronic hypoxia,and chronic anoxia in primary cortical neurons,neuroblastoma cells,and cerebral m icrovascular smooth muscle cells,respectively,in vitro.43—45However,this is the fi rst study,to our know ledge,demonstrating an acute loss of hippocampal ADAM 10 expression follow ing cerebral ischem iain vivo.Interestingly,E2 signaling has been recently linked w ith modulation of ADAM 10 in the brain.In fact,two green tea derivatives,(-)-epigallocatechin-3 gallate(EGCG)and octyl gallate,were recently reported to reduce Aβplaque load in transgenic AD mouse models via an ERα/PI3K/Aktsignaling mechanism,which led to maturation and increasedα-secretase activity of ADAM 10.29,46An additional study revealed that adm inistration of 100 mg/kg E2 to an ovariectom ized,D-galactose-injected rat model of AD led to elevation of ADAM 10,reduction of BACE1,and alleviation of spatial memory deficits.27The current study corroborates these findings by showing that low,Diestrus I levels of E2 are capable of preventing GCI-induced loss of hippocampal ADAM 10in vivo.Furthermore,our results expand upon these findings by demonstrating E2 regulation of ADAM 10 expression in w ild-type,non-transgenic rodents, suggesting that E2 may play a key role in endogenous nonamyloidogenic processing of APP in the hippocampus.It should be mentioned here that a single study which used a longer(4-month)ovariectomy period,observed an increase in ADAM 10 mRNA in the absence of ischem ia.28While the current study did not find an elevation of ADAM 10 expression in non-ischem ic LTED sham animals,it used a much shorter ovariectomy period(10 weeks).Thus,these results are not necessarily in disagreement.
The second novel finding of our study was evidence of a post-ischemic sw itch to amyloidogenic processing of APP in the hippocampal CA1 region follow ing LTED.In particular, we observed a loss of protein expression of bothα-secretases ADAM 10 and ADAM 17,an elevation of protein expression of theβ-secretase BACE1,and an increase in the C99/C83 protein ratio in the hippocampal CA1 of LTED females subjected to GCI.While neuronal expression of theα-secretase ADAM 10 has not been previously studied in the context of ischem iain vivo,neuronalexpression of ADAM 17,or TACE, has been demonstrated to be enhanced follow ing ischem ic preconditioning.47,48Since this suggests that up-regulation of ADAM 17/TACE is a neuroprotective mechanism responsible for the phenomenon of ischem ic tolerance,down-regulation of anα-secretase,like ADAM 17,in the hippocampus follow ing ischemic stress could be detrimental to neurons.As such,a marked decrease in expression of two neuroprotectiveα-secretases in LTED females after ischem ia(both ADAM 10 and ADAM 17)could partially explain the hippocampal hypersensitivity to GCI-induced cell loss observed in LTED females.4,49,50While exogenous E2 has been shown previously to modulate expression of both ADAM 10 and ADAM 17in vitroandin vivo,26,27,31this is the fi rst study to suggest that ovarian-derived E2 may promote non-amyloidogenic processing of APP follow ing ischem ic stress via modulation of α-secretase expression in hippocampalneuronsin vivo.Along w ith the significant increase in the amyloidogenic C99/C83 APP fragment ratio and the significant increase in BACE1 expression in the hippocampal CA1 of LTED females subjected to GCI,a robust loss of non-amyloidogenic ADAM 10 and ADAM 17 expression suggests that prolonged loss of ovarian E2 may promote a sw itch to amyloidogenic processing of APP in the eventof ischemia.This finding extends a recent report by our laboratory,which described a post-ischem ic sw itch to amyloidogenic processing of APP in the hippocampal CA3 region of LTED females,which becomes hypersensitive to both GCI and Aβneurotoxicity follow ing 10-week ovariectomy.4The current study demonstrates that this process also occurs in the critical hippocampal CA1 region of LTED females.Furthermore,it shows that E2 is capable of regulating two putativeα-secretases(ADAM 10 and ADAM 17) in addition to its known regulation of theβ-secretase BACE1. These additional findings are particularly important because they suggest that the post-ischem ic sw itch to amyloidogenic APP processing that occurs follow ing LTED is not regionspecific.A long these lines,it w ill be important for future studies to determ ine whether long-term ovariectomy only enhances stress-induced amyloidogenesis in the hippocampus or if this eventoccurs in other critical regions of the brain,such as the cerebral cortex.
The third major finding of the current study was an increased Aβload in the hippocampal CA1 of LTED females subjected to GCI.This observation corroborates the changes seen inα-secretase expression,β-secretase expression,and the C99/C83 ratio follow ing ischem ia in LTED females,suggesting that the post-ischem ic sw itch to amyloidogenic processing of APP does,in fact,enhance amyloidogenesis in the hippocampus of surgically menopausal females.Furthermore, this finding agrees w ith our previous study,which observed a sw itch to amyloidogenic APP processing and a resulting increase in Aβimmunoreactivity in the hippocampal CA3 region of LTED females follow ing GCI.4While notexam ined in this study,it is possible that a loss of E2 regulation of Aβclearance mechanisms could occur follow ing LTED as well. Considering that E2 has been shown to up-regulate both neprilysin and insulin-degrading enzyme,20—24as well as induce microglial phagocytosis of Aβ,51it would be interesting to determine whether long-term loss of ovarian E2 increases post-ischem ic hippocampal Aβload only through enhancing amyloidogenesis or whether ithinders the clearance of insoluble Aβas well.One concern is that premature surgicalmenopause,which is associated with a doubled lifetime risk of dementia,1alone may enhance neurotoxic Aβdeposition in the brain in the absence of ischem ia.However,neither the currentstudy nor our previously published work4found an increase of Aβin the hippocampus of LTED sham animals. Furthermore,an unrelated study found that the total hippocampal BACE1/ADAM 10 mRNA ratio,which reflects the status of amyloidogenic processing of APP,was unchanged in non-ischem ic females ovariectom ized for 4 months.28Together,these studies suggest that LTED alone does not promote a sw itch to amyloidogenic processing of APP and that an acute stressor is required for hippocampalamyloidogenesis to occur.
The fourth and final finding was a loss of E2 regulation of post-ischem ic changes in hippocampal ADAM 10,ADAM 17, BACE1,and PHF follow ing LTED.These results agree w ith our previous study,which found a loss of E2 regulation of BACE1 and PHF in the hippocampal CA3 region of LTED females after ischem ia.4Furthermore,it extends the aforementioned study to the critical CA1 region of the hippocampus and shows,for the fi rst time,that E2’s ability to regulate α-secretase expression is lost follow ing premature surgical menopause.Importantly,this finding is also in agreementw ith a grow ing body of literature that suggests low-dose E2 has a decreased ability to regulate neural factors follow ing longterm ovariectomy.4,49,50,52—54,55—57One important question is whether enhanced post-ischem ic development of AD-like neuropathology in a region critical for learning and memory would worsen neurocognitive outcome follow ing an ischem ic insult.Indeed,our colleagues found that ischemic LTED female rats,which displayed an increased Aβload in the CA3 region of the hippocampus,performed worse on the Morris water maze than their ischemic STED counterparts.4This suggests that the enhanced ischemia-induced AD-like neuropathology seen in LTED female rats may further impair neurocognitive functioning.
5.Conclusion
The current study provides evidence that prolonged loss of ovarian E2,through premature surgical menopause,could predispose the female hippocampus to development of AD-like neuropathology(increased hippocampal Aβand PHF)in the eventof ischem ic stress.This could occur due to the loss of E2’s ability to regulate post-ischem ic changes in AD-related proteins,such as the α- and β-secretases and the m icrotubule-associated protein tau.As such,these detrimental molecular changes could underlie the enhanced risk of dementia and mortality from neurological disorders observed in prematurely menopausal women.
Acknow ledgment
The research presented in this article was completed in partial fulfi llment of the MD/PhD dual degree requirements set forth for ELS atGeorgia Regents University and was supported by a Pre-doctoral Fellowship from the American Heart Association to ELS.(12PRE11530009)and a Research Grant from the National Institute of Neurological Disorders and Stroke, National Institutes of Health,USA to DWB(NS050730).
1.Rocca WA,Bower JH,Maraganore DM,Ahlskog JE,Grossardt BR,de Andrade M,et al.Increased risk of cognitive impairmentor dementia in women who underwent oophorectomy before menopause.Neurology2007;69:1074—83.
2.Rivera CM,GrossardtBR,Rhodes DJ,Rocca WA.Increased mortality for neurological and mental diseases follow ing early bilateraloophorectomy.Neuroepidemiology2009;33:32—40.
3.Rocca WA,Grossardt BR,Shuster LT.Oophorectomy,menopause,estrogen treatment,and cognitive aging:clinical evidence for a w indow of opportunity.Brain Res2011;1379:188—98.
4.Zhang QG,Wang RM,Scott E,Han D,Dong Y,Tu JY,et al.Hypersensitivity of the hippocampal CA3 region to stress-induced neurodegeneration and amyloidogenesis in a rat model of surgical menopause.Brain2013;136(Pt 5):1432—45.
5.Ram irez-Bermudez J.A lzheimer’s disease:critical notes on the history of a medical concept.Arch Med Res2012;43:595—9.
6.Defina PA,Moser RS,Glenn M,Lichtenstein JD,Fellus J.Alzheimer’s disease clinical and research update for health care practitioners.J Aging Res2013;2013:207178.
7.Zheng H,Koo EH.Biology and pathophysiology of the amyloid precursor protein.Mol Neurodegener2011;6:27.http://dx.doi.org/10.1186/1750-1326-6-27.
8.Willnow TE,Petersen CM,Nykjaer A.VPS10P-domain receptors—regulators of neuronal viability and function.Nat RevNeurosci2008;9:899—909.
9.De Strooper B,Vassar R,Golde T.The secretases:enzymes w ith therapeutic potential in Alzheimer disease.Nat Rev Neurol2010;6:99—107.
10.Pike CJ,Carroll JC,Rosario ER,Barron AM.Protective actions of sex steroid hormones in Alzheimer’s disease.Front Neuroendocrinol2009;30:239—58.
11.Hogervorst E,Williams J,Budge M,Riedel W,Jolles J.The nature of the effect of female gonadal hormone replacement therapy on cognitive function in post-menopausal women:a meta-analysis.Neuroscience2000;101:485—512.
12.Brann DW,Dhandapani K,Wakade C,Mahesh VB,Khan MM.Neurotrophic and neuroprotective actions of estrogen:basic mechanisms and clinical implications.Steroids2007;72:381—405.
13.Paganini-Hill A,Henderson VW.Estrogen replacement therapy and risk of Alzheimer disease.Arch Intern Med1996;156:2213—7.
14.Fox M,Berzuini C,Knapp LA.Cumulative estrogen exposure,number of menstrual cycles,and Alzheimer’s risk in a cohort of British women.Psychoneuroendocrinology2013;38:2973—82.
15.Simpkins JW,Wen Y,Perez E,Yang S,Wang X.Role of nonfeminizing estrogens in brain protection from cerebral ischem ia:an animalmodel of A lzheimer’s disease neuropathology.AnnNYAcadSci2005;1052:233—42.
16.Zhang QG,Wang R,Khan M,Mahesh V,Brann DW.Role of Dickkopf-1, an antagonist of the Wnt/beta-catenin signaling pathway,in estrogeninduced neuroprotection and attenuation of tau phosphorylation.J Neurosci2008;28:8430—41.
17.Shah RD,Anderson KL,Rapoport M,Ferreira A.Estrogen-induced changes in the m icrotubular system correlate w ith a decreased susceptibility of aging neurons to beta amyloid neurotoxicity.Mol Cell Neurosci2003;24:503—16.
18.Marin R,Ram irez C,Morales A,Gonzalez M,Alonso R,Diaz M. Modulation of Abeta-induced neurotoxicity by estrogen receptor alpha and other associated proteins in lipid rafts.Steroids2008;73:992—6.
19.Yue X,Lu M,Lancaster T,Cao P,Honda S,Staufenbiel M,et al.Brain estrogen defi ciency accelerates Abeta plaque formation in an A lzheimer’s disease animal model.ProcNatl AcadSci USA2005;102:19198—203.
20.Li R,He P,Cui J,Staufenbiel M,Harada N,Shen Y.Brain endogenous estrogen levels determ ine responses to estrogen replacement therapy via regulation of BACE1 and NEP in female A lzheimer’s transgenic m ice.Mol Neurobiol2013;47:857—67.
21.Jayaraman A,Carroll JC,Morgan TE,Lin S,Zhao L,Arimoto JM,etal. 17beta-estradiol and progesterone regulate expression of beta-amyloid clearance factors in primary neuron cultures and female rat brain.Endocrinology2012;153:5467—79.
22.Liang K,Yang L,Yin C,Xiao Z,Zhang J,Liu Y,etal.Estrogen stimulates degradation of beta-amyloid peptide by up-regulating neprilysin.J Biol Chem2010;285:935—42.
23.Xiao ZM,Sun L,Liu YM,Zhang JJ,Huang J.Estrogen regulation of the neprilysin gene through a hormone-responsive element.J Mol Neurosci2009;39:22—6.
24.Huang J,Guan H,Booze RM,Eckman CB,Hersh LB.Estrogen regulates neprilysin activity in rat brain.Neurosci Lett2004;367:85—7.
25.Sambamurti K,Kinsey R,Maloney B,Ge YW,LahiriDK.Gene structure and organization of the human beta-secretase(BACE)promoter.FASEB J2004;18:1034—6.
26.Nord LC,Sundqvist J,Andersson E,Fried G.Analysis of oestrogen regulation of alpha-,beta-and gamma-secretase gene and protein expression in cultured human neuronal and glial cells.Neurodegener Dis2010;7:349—64.
27.Zhang X,Wang J,Xing Y,Gong L,Li H,Wu Z,et al.Effects of ginsenoside Rg1 or 17beta-estradiol on a cognitively impaired,ovariectom ized rat model of A lzheimer’s disease.Neuroscience2012;220:191—200.
28.Anukulthanakorn K,Malaivijitnond S,Kitahashi T,Jaroenporn S, Parhar I.Molecular events during the induction of neurodegeneration and memory loss in estrogen-defi cient rats.GenCompEndocrinol2013;181:316—23.
29.Fernandez JW,Rezai-Zadeh K,Obregon D,Tan J.EGCG functions through estrogen receptor-mediated activation of ADAM 10 in the promotion of non-amyloidogenic processing of APP.FEBSLett2010;584:4259—67.
30.Sun M,Zhou T,Zhou L,Chen Q,Yu Y,Yang H,et al.Formononetin protects neurons against hypoxia-induced cytotoxicity through upregulation of ADAM 10 and sAbetaPPalpha.JAlzheimersDis2012;28:795—808.
31.Nadadur RD,Umar S,Wong G,Eghbali M,Iorga A,Matori H,et al. Reverse right ventricular structural and extracellular matrix remodeling by estrogen in severe pulmonary hypertension.J Appl Physiol(1985)2012;113:149—58.
32.Zhang QG,Tian H,Li HC,Zhang GY.Antioxidant N-acetylcysteine inhibits the activation of JNK3 mediated by the GluR6-PSD95-MLK3 signaling module during cerebral ischem ia in rat hippocampus.Neurosci Lett2006;408:159—64.
33.Pulsinelli WA,Brierley JB.A new model of bilateral hem ispheric ischemia in the unanesthetized rat.Stroke1979;10:267—72.
34.Pulsinelli WA,Buchan AM.The four-vesselocclusion ratmodel:method for complete occlusion of vertebral arteries and control of collateral circulation.Stroke1988;19:913—4.
35.Wen Y,Yang S,Liu R,Simpkins JW.Transient cerebral ischemia induces site-specific hyperphosphorylation of tau protein.BrainRes2004;1022:30—8.
36.Wen Y,Yang S,Liu R,Brun-Zinkernagel AM,Koulen P,Simpkins JW. Transient cerebral ischemia induces aberrantneuronal cell cycle re-entry and A lzheimer’s disease-like tauopathy in female rats.J Biol Chem2004;279:22684—92.
37.Wen Y,Yang SH,Liu R,Perez EJ,Brun-ZinkernagelAM,Koulen P,etal. Cdk5 is involved in NFT-like tauopathy induced by transient cerebral ischem ia in female rats.Biochim Biophys Acta2007;1772:473—83.
38.Pluta R,Jablonski M,Ulamek-Koziol M,Kocki J,Brzozowska J, Januszewski S,et al.Sporadic A lzheimer’s disease begins as episodes of brain ischemia and ischemically dysregulated Alzheimer’s disease genes.Mol Neurobiol2013;48:500—15.
39.Epis R,Marcello E,Gardoni F,Vastagh C,Malinverno M,Balducci C, et al.Blocking ADAM 10 synaptic trafficking generates a model of sporadic Alzheimer’s disease.Brain2010;133:3323—35.
40.Schroeder A,Fahrenholz F,Schm itt U.Effect of a dom inant-negative form of ADAM 10 in a mouse model of Alzheimer’s disease.J Alzheimers Dis2009;16:309—14.
41.Gil-Bea F,Akterin S,Persson T,Mateos L,Sandebring A,Avila-Carino J, etal.Thioredoxin-80 is a productof alpha-secretase cleavage that inhibits amyloid-beta aggregation and is decreased in Alzheimer’s disease brain.EMBO Mol Med2012;4:1097—111.
42.Pietri M,Dakowski C,Hannaoui S,A lleaume-Butaux A,Hernandez-Rapp J,Ragagnin A,et al.PDK1 decreases TACE-mediated alpha-secretase activity and promotes disease progression in prion and Alzheimer’s diseases.Nat Med2013;19:1124—31.
43.Marshall AJ,Rattray M,Vaughan PF.Chronic hypoxia in the human neuroblastoma SH-SY5Y causes reduced expression of the putative alphasecretases,ADAM 10 and TACE,w ithout altering their mRNA levels.Brain Res2006;1099:18—24.
44.Lee PH,Hwang EM,Hong HS,Boo JH,Mook-Jung I,Huh K.Effectof ischem ic neuronal insults on amyloid precursor protein processing.Neurochem Res2006;31:821—7.
45.Auerbach ID,Vinters HV.Effects of anoxia and hypoxia on amyloid precursor protein processing in cerebral microvascular smooth muscle cells.J Neuropathol Exp Neurol2006;65:610—20.
46.Zhang SQ,Sawmiller D,LiS,Rezai-Zadeh K,Hou H,Zhou S,etal.Octyl gallate markedly promotes anti-amyloidogenic processing of APP through estrogen receptor-mediated ADAM 10 activation.PLoSOne2013;8:e71913.http://dx.doi.org/10.1371/journal.pone.0071913.
47.Cardenas A,Moro MA,Leza JC,O’Shea E,Davalos A,Castillo J,et al. Upregulation of TACE/ADAM 17 after ischem ic preconditioning is involved in brain tolerance.JCereb Blood Flow Metab2002;22:1297—302.
48.Pradillo JM,Romera C,Hurtado O,Cardenas A,Moro MA,Leza JC,etal. TNFR1 upregulation mediates tolerance after brain ischem ic preconditioning.J Cereb Blood Flow Metab2005;25:193—203.
49.Zhang QG,Han D,Wang RM,Dong Y,Yang F,Vadlamudi RK,etal.C term inus of Hsc70-interacting protein(CHIP)-mediated degradation of hippocampal estrogen receptor-alpha and the critical period hypothesis of estrogen neuroprotection.ProcNatlAcadSciUSA2011;108:E617—24.
50.Zhang QG,Raz L,Wang R,Han D,De Sevilla L,Yang F,etal.Estrogen attenuates ischem ic oxidative damage via an estrogen receptor alphamediated inhibition of NADPH oxidase activation.JNeurosci2009;29:13823—36.
51.Li R,Shen Y,Yang LB,Lue LF,Finch C,Rogers J.Estrogen enhances uptake of amyloid beta-protein by m icroglia derived from the human cortex.J Neurochem2000;75:1447—54.
52.Ham ilton RT,Rettberg JR,Mao Z,To J,Zhao L,Appt SE,et al.Hippocampal responsiveness to 17beta-estradiol and equol after long-term ovariectomy:implication for a therapeutic w indow of opportunity.Brain Res2011;1379:11—22.
53.Ding F,Yao J,Zhao L,Mao Z,Chen S,Brinton RD.Ovariectomy induces a shift in fuel availability and metabolism in the hippocampus of the female transgenic modelof fam ilial A lzheimer’s.PLoS One2013;8:e59825. http://dx.doi.org/10.1371/journal.pone.0059825.
54.Bohacek J,Daniel JM.The benefi cial effects of estradiol on attentional processes are dependent on tim ing of treatment initiation follow ing ovariectomy in m iddle-aged rats.Psychoneuroendocrinology2010;35:694—705.
55.Raz L,Zhang QG,Han D,Dong Y,De Sevilla L,Brann DW.Acetylation of the pro-apoptotic factor,p53 in the hippocampus follow ing cerebral ischem ia and modulation by estrogen.PLoS One2011;6:e27039.http:// dx.doi.org/10.1371/journal.pone.0027039.
56.Scott EL,Zhang QG,Han D,Desai BN,Brann DW.Long-term estrogen deprivation leads to elevation of Dickkopf-1 and dysregulation of Wnt/ beta-Catenin signaling in hippocampal CA1 neurons.Steroids2013;78:624—32.
57.Suzuki S,Brown CM,Dela Cruz CD,Yang E,Bridwell DA,Wise PM. Tim ing of estrogen therapy after ovariectomy dictates the efficacy of its neuroprotective and antiinflammatory actions.Proc Natl Acad Sci U S A2007;104:6013—8.
Received 24 January 2014;revised 1 April 2014;accepted 5 April 2014
*Corresponding author.
E-mail address:dbrann@gru.edu(D.W.Brann)
Peer review under responsibility of Shanghai University of Sport
2095-2546/$-see front matter CopyrightⒸ2014,Shanghai University of Sport.Production and hosting by Elsevier B.V.A ll rights reserved. http://dx.doi.org/10.1016/j.jshs.2014.04.003
 Journal of Sport and Health Science2014年3期
Journal of Sport and Health Science2014年3期
- Journal of Sport and Health Science的其它文章
- Women’s health in exercise and aging:What do we know?
- Why women see differently from the way men see?A review of sex differences in cognition and sports
- Sex differences in exercise and drug addiction:A m ini review of animal studies
- Women and exercise in aging
- Effects of carbohydrate supplements on exercise-induced menstrual dysfunction and ovarian subcellular structural changes in rats
- Exercise training and antioxidant supplementation independently improve cognitive function in adult male and female GFAP-APOE m ice