
VEGF在EAE大鼠血清中的表达及意义
2014-03-18 03:38杨慧敏蹇顺海黄一凡李祖茂
川北医学院学报 2014年5期
杨慧敏,蹇顺海,黄一凡,李祖茂
(川北医学院基础医学院病理学教研室,四川 南充 637000)
多发性硬化(multiple sclerosis,MS)是以中枢神经系统白质炎性脱髓鞘病变为主要特点的自身免疫性疾病,具有较高的致残率。近几年的研究提出了自身免疫、病毒感染、遗传倾向等综合作用的多因素病因学说,但其病因和发病机制至今仍未完全明确。目前多采用敏感动物制作与MS发病极为相似的疾病模型即实验性自身免疫性脑脊髓炎(experimental autoimmune encephalomyelitis,EAE)模型对MS进行研究。有国外研究者[1]在EAE疾病进展中观察到血管生成现象,且与临床表现保持平行,但其与髓鞘炎性破坏的具体关系目前尚未见相关研究。本研究拟通过复制大鼠EAE模型,观察与血管生成有重要关系的血管内皮生长因子(vascular endothelial growth factor,VEGF)的表达,探讨其与EAE发生发展的关系及其临床意义。
1 材料与方法
1.1 材料
1.1.1 动物与分组 健康雌性Wistar大鼠30只,体重180~220 g,随机分组为模型组20只及对照组10只;豚鼠1只,体重350 g,均由川北医学院动物实验中心提供。
1.1.2 主要试剂 卡介苗冻干粉、百日咳菌液购自中国药品生物制品检定所,羊毛脂、液体石蜡购自南充药业集团;固蓝(luxol fast blue,LFB)购自SIGMA公司;大鼠VEGF、IL-4、IL-10及IFN-γ ELISA试剂盒购自欣博盛生物科技有限公司,大鼠髓鞘碱性蛋白(myelin basic protein,MBP)ELISA试剂盒购自武汉优尔生科技股份有限公司。
1.2 方法
1.2.1 动物模型制作 羊毛脂20 g加入液体石蜡40 mL混匀,灭菌后每毫升加入卡介苗4~10 mg混匀即成完全弗氏佐剂,将豚鼠全脑及脊髓制成50%脑脊髓匀浆,与完全弗氏佐剂1∶1充分混匀即得到完全抗原。模型组20只,麻醉消毒后于四肢足垫内注射完全抗原0.4 mL/只,同时腹部皮内注射百日咳菌液0.1 mL(约含活菌3×109),对照组10只,麻醉消毒后仅足垫注射等量生理盐水。
1.2.2 模型判定 每日称重,并参照Hooper等[2]的方法进行评分:0分:不发病;1分:竖毛,尾无力;2分:尾瘫;3分:尾瘫及后肢无力;4分:尾瘫及不完全后肢瘫;5分:完全后肢瘫;6分:四肢瘫;7分:濒死或死亡。
1.2.3 取材 将模型组大鼠于发病高峰时(症状连续3 d无加重或评分6~7分时)处死,对照组大鼠于模型组全部处死同时处死。深度麻醉后,抽取右心房血液备用,用小号静脉留置针经左心室穿刺,再用生理盐水推注冲洗及4%多聚甲醛灌注固定后取全脑、脊髓。
1.2.4 病理学观察 分离的大鼠全脑及脊髓用4%多聚甲醛固定24~48 h后行石蜡包埋切片及HE、LFB染色。
1.2.5 用ELISA检测各因子在各组大鼠血清中的浓度 大鼠血液在促凝管中静置30 min,3 000 r/min离心10 min分离血清,按试剂盒说明书用ELISA法检测血清中VEGF、IL-4、IL-10、IFN-γ及MBP浓度。
1.3 统计学分析
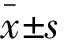
2 结果
2.1 EAE发病情况及病理学观察
2.1.1 发病情况 全部模型组大鼠于免疫后(13.33±1.88) d开始出现症状,发病率为100%。具体表现为进食及活动减少,体重下降,进而尾部无力、后肢无力、大小便失禁、前肢无力等,观察期间未出现死亡者。模型组发病高峰临床评分为(4.4±1.35)分。对照组进食、活动情况良好,体重无减轻,无一发病。
2.1.2 病理学观察 (1)HE染色:EAE主要的光镜下形态学表现为炎症性改变,主要表现为脑脊髓炎性水肿,血管扩张充血,以淋巴细胞为主的炎细胞浸润。实验组于发病高峰期取材、染色后观察发现,大鼠的大脑、小脑及脊髓中有大量炎细胞浸润,并可围绕血管呈“血管袖套样”改变,以脊髓及小脑病变最为明显,主要集中于脊髓表面蛛网膜下腔及近表面的白质内,炎细胞浸润明显区由于水肿,组织间隙增宽。对照组大鼠中枢神经系统未见明显改变(图1)。(2)LFB染色:LFB可将正常髓鞘染成蓝色,EAE病理改变为炎性髓鞘脱失,故脱髓鞘区不被LFB染色或只见髓鞘碎片。模型组大鼠发病高峰时中枢神经系统在炎细胞浸润明显区可见明显髓鞘脱失,呈空泡状改变,病变较轻区域可见髓鞘碎片(图2)。对照组大鼠中枢神经系统未见明显脱髓鞘病变。
2.2 大鼠血清中各细胞因子表达检测
模型组大鼠发病高峰时血清中VEGF、IFN-γ及MBP水平较对照组明显升高,IL-4水平较对照组降低,差异有统计学意义(P<0.05)。模型组血清IL-10水平与对照组相比较,差异无统计学意义(表1)。
表1 大鼠血清中各细胞因子表达情况及组间比较

VEGFIFN-γIL-4IL-10MBP模型组29.98±14.8420.71±12.3482.88±38.4211.53±8.5422.47±13.34对照组19.70±10.447.91±4.48144.91±30.155.24±1.154.69±2.65P值0.0430.0220.0190.2320.000t值2.1232.241-2.6161.2434.715
2.3 相关性分析
模型组大鼠中,临床症状评分与VEGF水平呈显著正相关(P<0.01),与IFN-γ及MBP水平亦呈正相关,与IL-4水平呈负相关,相关性存在统计学差异(P<0.05),与IL-10水平无明显相关性(表2)。模型组大鼠血清中VEGF水平与IFN-γ及MBP水平呈正相关,与IL-4水平呈负相关,相关性存在统计学差异(P<0.05),与IL-10水平无明显相关性(表3)。

表2 模型组临床评分与各细胞因子表达的相关性
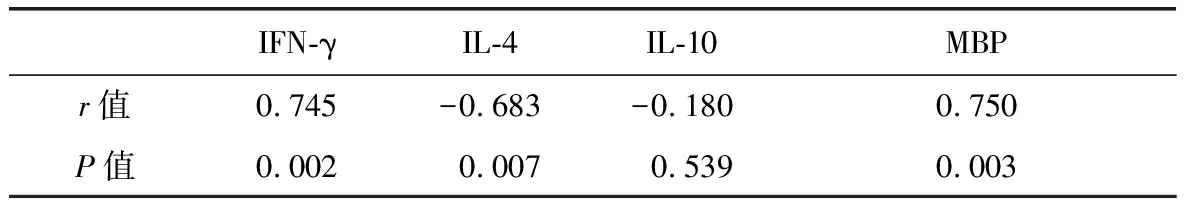
表3 模型组VEGF与其余各细胞因子表达的相关性
3 讨论
MS的确切发病机制仍不十分清楚,目前多认为是T淋巴细胞介导的炎性反应,细胞因子在其中起着重要作用。研究表明,MS患者中存在Thl/Th2类细胞因子的失衡,Th1细胞主要分泌促炎症性细胞因子(如IL-2、IL-12、IL-17、IFN-γ、TNF-α等),可激活巨噬细胞,介导迟发性变态反应诱发EAE;Th2细胞主要分泌抑炎症性细胞因子(如IL-4、IL-5、IL-10、IL-6等),可促进B细胞分化成熟,产生抗体,增强抗体介导的免疫应答,抑制或逆转EAE的发生。细胞因子在EAE发病及病情进展中起着重要而复杂的调节作用[3-5]。
EAE作为MS经典动物模型,其病理改变与MS相似[6],主要是脑和脊髓实质内小血管周围和蛛网膜下腔内有大量淋巴细胞浸润。淋巴细胞在血管周围形成典型的袖套状改变,病变部位髓鞘崩解、脱失,神经细胞变性、坏死,上述改变亦可以在我们的实验中观察到。研究发现[7-8],EAE急性损害还包括血脑屏障(blood brain barrier,BBB)分解和病灶内毛细血管内皮细胞上黏附分子上调,提示血管通透性改变和内皮细胞的活化是MS发病机制中的重要部分。Th1细胞因子在其发病中的作用可能是通过上调病灶内毛细血管内皮细胞上黏附分子,使活化的T细胞得以穿过BBB进入脑实质中[9],另外Th1细胞因子还可诱导基质金属蛋白酶(matrix metalloproteinase,MMPs)表达,促进BBB破坏和继发脑组织损伤。
VEGF具有促进新生血管生成、增加血管通透性和促进单核细胞游走等功能[10]。而局部组织的缺血和缺氧可诱导VEGF的产生,与其相应的受体结合,激活内皮细胞,促进内皮细胞的增殖和迁移、基底膜的降解及表达某些整合素。研究者发现,血清VEGF水平在MS患者急性复发期较健康对照组或MS缓解期有所增加[11];Proescholdt等[7]研究发现,MS脱髓鞘损害中VEGF表达有显著增高,且病灶内血管数目增高与临床及病理表现(即炎症细胞浸润和脱髓鞘)进展保持平行。
在本研究中,我们成功复制了大鼠EAE模型,模型组大鼠均不同程度出现由尾部开始的运动功能障碍症状。大多数EAE研究采用Kono’s 5分法[12]进行症状评分,但5分的分类相对粗略而不利于统计学分析,为了对症状的观察和评价更加敏感和精细,我们采用Hooper’s改良后的7分法作为EAE神经功能损伤评分法,模型组发病高峰临床评分为(4.4±1.35)分。因欲观察EAE大鼠症状最明显时病理表现与细胞因子表达的关系,我们在发病高峰时获取大鼠的组织及血清检材。
在我们的研究中发现,VEGF在EAE大鼠血清中表达较对照组有明显增高,且与动物模型发病临床评分呈显著正相关,提示VEGF的增高很可能与EAE病变的进展有关;同时,VEGF与血清IFN-γ及MBP水平升高相一致(前者表示促炎因子的水平,后者可表示髓鞘损伤的程度),与IL-4水平(可表示抑炎因子的水平)升高呈负相关,相关性检验具有统计学意义。与对照组相比,IL-4水平在模型组中显著下降,IFN-γ水平显著增高;IFN-γ在模型组大鼠血清中的水平与其临床评分呈正相关,IL-4水平与临床评分呈负相关,均具有统计学意义,与前人研究结果相符[3]。
综合上述发现,我们认为,EAE时血清中增高的VEGF可以是炎症区的血管内皮细胞释放及巨噬细胞产生[13],在其病变进展中,VEGF可能通过活化炎区的血管内皮细胞产生作用:(1)诱导活化的血管内皮细胞分泌MMPs,降解细胞外基质,促进炎细胞浸润;(2)使血管通透性增高,局部趋化浸润的炎细胞增多,释放促炎因子,一方面直接参与炎症过程,另一方面使血管内皮细胞上黏附分子表达上调而促进炎细胞浸润,进一步破坏BBB;(3)诱导炎区新生血管形成,新生血管的内皮细胞不成熟,基底膜不完整,通透性大,炎细胞易于通过而进入病区。VEGF通过上述可能的机制与炎细胞释放的细胞因子及调高的MMPs共同作用或诱导新生血管形成,致BBB免疫屏障受损,炎细胞到达CNS白质参与炎症反应,导致髓鞘破坏。这可能为MS的治疗打开新的思路,即抑制VEGF,使与之相关的炎症促进因素得到控制,从而减轻炎症破坏。
当然,MS及其模型EAE的发病机制目前认识不足,VEGF与之病变进展的研究也才刚开始,对于其中的确切联系及抑制VEGF对MS及EAE是否有效还有待进一步研究。
【参考文献】
[1]Kirk SL,Karlik SJ.VEGF and vascular changes in chronic neuroinflammation[J].J Autoimmun,2003,21(4):353-363.
[2]Hooper DC,Spitsin S,Kean RB,et al.Uric acid,a natural scavenger of peroxynitrite,in experimental allergic encephalomyelitis and multiple sclerosis[J].Proc Natl Acad Sci USA,1998,95(2):675-680.
[3]Amedei A,Prisco D,D’Elios MM.Multiple sclerosis:the role of cytokines in pathogenesis and in therapies[J].Int J Mol Sci,2012,13(10):13438-13460.
[4]Hamann I,Zipp F,Infante-Duarte C.Therapeutic targeting of chemokine signaling in Multiple Sclerosis[J].J Neurol Sci,2008,274(1-2):31-38.
[5]Comabella M,Khoury SJ.Immunopathogenesis of multiple sclerosis[J].Clin Immunol,2012,142(1):2-8.
[6]朱振国,王新施,陈艳艳,等.3种大鼠实验性自身免疫性脑脊髓炎模型的差异[J].医学研究杂志,2013,42(2):34-37.
[7]Proescholdt MA,Jacobson S,Tresser N,et al.Vascular endothelial growth factor is expressed in multiple sclerosis plaques and can induce inflammatory lesions in experimental allergic encephalomyelitis rats[J].J Neuropathol Exp Neurol,2002,61(10):914-925.
[8]Argaw AT,Asp L,Zhang J,et al.Astrocyte-derived VEGF-A drives blood-brain barrier disruption in CNS inflammatory disease[J].J Clin Invest,2012,122(7):2454-2468.
[9]O’Connor RA,Prendergast CT,Sabatos CA,et al.Cutting edge:Th1 cells facilitate the entry of Th17 cells to the central nervous system during experimental autoimmune encephalomyelitis[J].J Immunol,2008,181(6):3750-3754.
[10]Carvalho JF,Blank M,Shoenfeld Y.Vascular endothelial growth factor(VEGF)in autoimmune diseases[J].J Clin Immunol,2007,27(3):246-256.
[11]Su JJ,Osoegawa M,Matsuoka T,et al.Upregulation of vascular growth factors in multiple sclerosis:correlation with MRI findings[J].J Neurol Sci,2006,243(1):21-30.
[12]Urban JL,Kumar V,Kono DH,et al.Restricted use of T cell receptor V genes in murine autoimmune encephalomyelitis raises possibilities for antibody therapy[J].Cell,1988,54(4):577-592.
[13]Jeon SH,Chae BC,Kim HA,et al.Mechanisms underlying TGF-beta 1-induced expression of VEGF and Flk-1 in mouse macrophages and their implications for angiogenesis[J].J Leukoc Biol,2007,81(2):557-566.
猜你喜欢
中华耳科学杂志(2022年1期)2022-11-24
生物化学与生物物理进展(2022年11期)2022-03-02
现代临床医学(2021年4期)2021-07-31
中国眼镜科技杂志(2019年9期)2019-11-11
党的生活(黑龙江)(2018年9期)2018-10-17
益寿宝典(2018年1期)2018-01-27
安徽医科大学学报(2016年12期)2017-01-15
中西医结合心脑血管病杂志(2016年20期)2016-03-01
中国病理生理杂志(2015年8期)2015-12-21
医学研究杂志(2015年12期)2015-06-10
