
我院药物临床试验知情同意书设计及签署情况分析
2014-03-17 08:46郭晋敏赵稳华康长清
中国医药导报 2014年12期
郭晋敏 张 莉 舒 鹤 赵稳华 康长清
济南军区总医院药剂科,山东济南250031
我院药物临床试验知情同意书设计及签署情况分析
郭晋敏 张 莉 舒 鹤 赵稳华 康长清
济南军区总医院药剂科,山东济南250031
目的分析济南军区总医院(以下简称“我院”)药物临床试验知情同意书的设计及签署情况。方法制订知情同意书设计及签署要素标准,调查我院58项药物临床试验知情同意书的设计及2042份知情同意书的签署中各要素的出现率,并对出现率低的要素进行深入分析。结果知情同意书的设计及签署基本符合《药物临床试验质量管理规范》的要求,但仍存在要素缺失及签署或修改不规范问题。国内项目的设计要素中,各缺失要素的出现率均低于国际多中心,出现率较低的有入选/排除标准(13.7%比71.4%)、试验保险及赔偿(7.8%比57.1%)、伦理委员会的联系人及联系方式(3.9%比85.7%)、知情同意书版本号及版本日期(41.2%比100.0%)。签署要素中以下容易被忽视:受试者联系方式、研究者联系方式未填写或填写固定电话、法定代理人代签时未注明受试者姓名及与受试者的关系,进一步分析发现,部分签署的缺失源于签字页项目设计不完整。结论针对发现问题提出对策,规范知情同意书的管理,促进我院药物临床试验的规范开展。
药物临床试验;知情同意书;设计;签署
药物临床试验是确证新药在人体内有效性和安全性必不可少的步骤。为了规范临床试验过程,确保其科学可靠,保护受试者的权益并保障其安全,原国家食品药品监督管理局(SFDA)在2003年发布了《药物临床试验质量管理规范》[1],在其中第三章第14、15条中对知情同意做出了明确的规定。知情同意是临床试验中保障受试者权益的主要措施,在开始试验之前,研究者必须向受试者知情告知,并签署知情同意书(特殊情况下可由法定代理人签署)。
尽管药物临床试验研究者均按规定执行了知情同意,伦理委员会和机构办公室对其也进行了监管,但在实践中仍存在很多问题[2-4]。为了解济南军区总医院(以下简称“我院”)知情同意的现况,本研究回顾性调查2006年1月~2012年6月已完成的58个药物临床试验中的知情同意书,从知情同意书的设计及签署情况方面进行分析,旨在发现我院知情同意工作中存在的问题,制订相应对策,加强管理,保障高水平新药临床研究工作的顺利开展。
1 资料与方法
1.1 一般资料
我院2006年1月~2012年6月期间已完成的Ⅱ、Ⅲ期药物临床试验58项,知情同意书共2042份。
1.2 方法
参考国内外指南[5-6],制定知情同意书的设计及签署要素标准。对照设计要点,调研各项目知情同意书中每条要素的出现率;对照签署要点,调研每份知情同意书中签署要素的出现率。根据调查结果,分析知情同意工作的现状及存在的问题。
2 结果
2.1 知情同意书设计要素标准
结合国内外相关指南和原则制订[5-6],具体如下:①研究背景;②项目目的;③试验流程;④试验期限;⑤入选/排除标准;⑥试验可能的受益;⑦试验分组情况;⑧试验可能的风险;⑨试验药物及检查免费;⑩替代治疗方法;试验保密及保护受试者隐私;试验保险及赔偿;受试者的权利告知(自愿、自由参加及退出);重新获得知情同意;受试者签名及日期;受试者联系方式;研究者签名及日期;研究者联系方式;法定代理人签名及日期;法定代理人与受试者的关系;伦理委员会的联系人及联系方式;知情同意书的版本号及版本日期。其中,1~4、6~8、 11~13项为我国《药物临床试验质量管理规范》规定知情同意书需告知的要素。
2.2 知情同意书的设计完整性
将项目按照申办者性质分为国内(51项)和国际(7项),分别对知情同意书设计要素进行综合分析,国内与国际知情同意书基本涵盖1、2、4、6~11、13~15、17项(出现率>95%),除12项(试验保险及赔偿),知情同意书的设计基本符合药品临床试验管理规范的要求。
对缺失要素的情况进行分析,结果详见表1。由表1可见,国内知情同意书要素中3、16、18项出现率较高,为82.3%、64.7%、74.5%,国际的出现率分别为100.0%、57.1%、100.0%。国内要素5(13.7%)、12(7.8%)、19(49.0%)、20(23.5%)、21(3.9%)、22项(41.2%)出现率较低,而国际出现率分别为71.4%、57.1%、85.7%、57.1%、85.7%、100.0%。国内各缺失要素的出现率均低于国际多中心。
2.3 知情同意书签署要素标准
结合实践与经验制订如下:①受试者签名及日期;②受试者联系方式;③研究者签名及日期;④研究者联系方式;⑤法定代理人签名及日期;⑥注明法定代理人与受试者的关系;⑦签署早于试验;⑧受试者与研究者签署日期相同。备注:2项只适用于受试者本人签署的知情同意书,5、6项只适用于法定代理人代签的知情同意书。其中,1、3、5、7、8项为我国《药物临床试验质量管理规范》规定的签署要素标准。

表1 知情同意书缺失要素分析[n(%)]
2.4 知情同意书的签署完整性
对8项签署标准进行统计,见表2。其中,1、3、5、7、8项基本签署完整规范,符合《药物临床试验质量管理规范》的要求,但以下要素容易被忽视,包括2项(受试者联系方式)、4项(研究者联系方式)和法定代理人代签时的1项(受试者姓名)、6项(注明法定代理人与受试者的关系)。对以上4项联合设计和签署角度进行分析发现,设计第2项并签署的占47.41%,未设计但签署的占8.89%,设计但未签署的占24.50%,未设计且未签署的占43.70%(图1);法定代理人代签时,均设计了第1项,签署的占65.43%(图2);法定代理人代签时,设计第6项并注明的占29.63%,未设计但注明占21.60%,设计但未注明的占0.62%,未设计且未注明的占48.15%(图3);设计第4项且签署手机号码的占53.57%,设计且签署固定电话的占24.58%,设计但未签署的占4.55%,未设计且未签署的占17.29%(图4)。
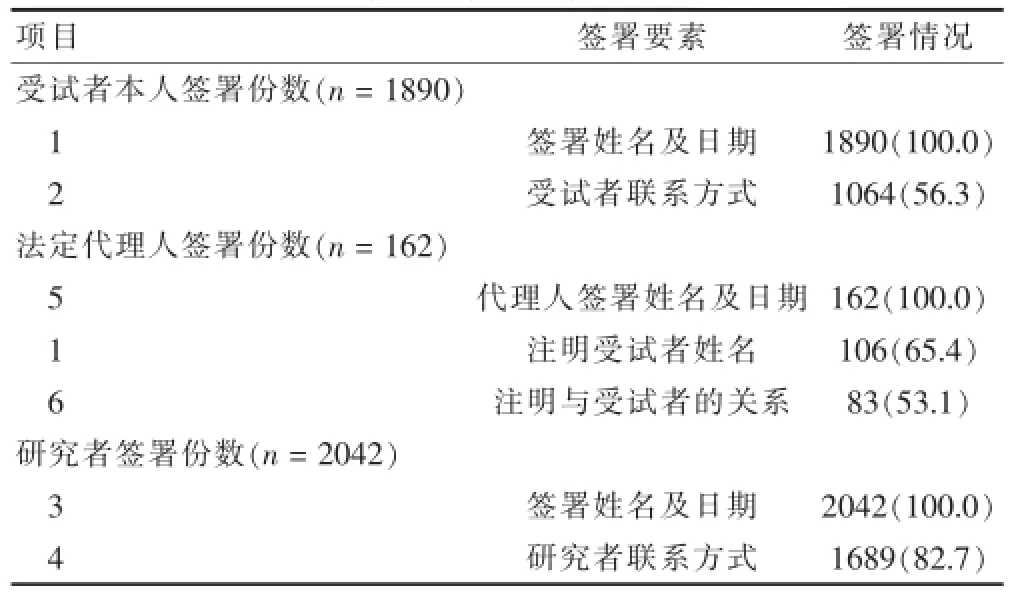
表2 知情同意书签署情况分析[n(%)]

图1 受试者联系方式签署情况分析

图2 代理人代签时受试者姓名注明情况分析
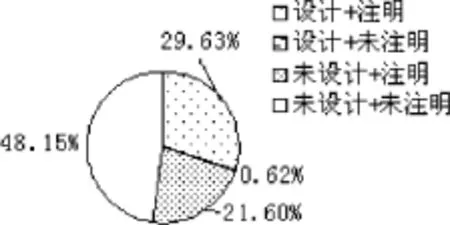
图3 代理人与受试者关系注明情况分析
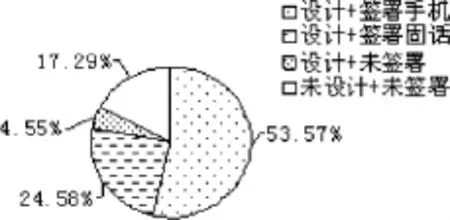
图4 研究者联系方式签署情况分析
2.5 知情同意书的签署修改规范性
知情同意书规范性修改应使用杠改,保持原记录可见,在旁边填写正确记录,并签署修改人的姓名(或拼音缩写)及修改日期。本次调研中知情同意书签署修改的规范率较低,受试者及研究者分别为10.6%和29.4%(图5)。
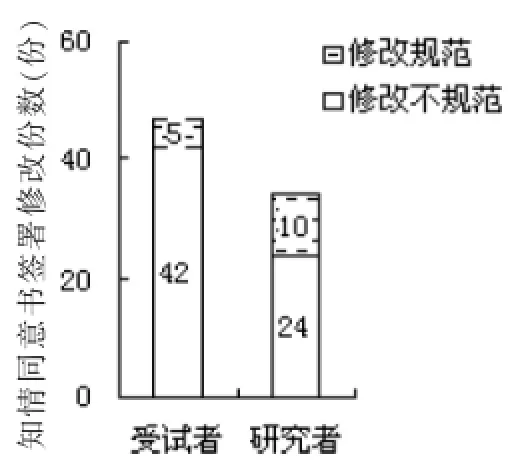
图5 知情同意书中签署修改情况分析
3 讨论
3.1 国际多中心知情同意书设计完整性及质量高于国内本次调研中,知情同意书设计缺失要素国际有6项,国内有9项,且国内缺失要素的出现率均低于国际,总体上国际知情同意书设计完整性高于国内。国内外知情同意书设计质量存在显著差异的要素有:①国际多中心临床试验更重视受试者的权益保护,知情同意书中多设计伦理委员会的联系人及电话,而国内多中心临床试验该部分意识则较薄弱。②规范的知情同意书应注明版本号及日期,这一信息有助于确认所签署的知情同意书是获得伦理委员会批准的正确版本的知情同意书[4]。国际知情同意书均注明版本号及版本日期,而国内该项出现率不足50%。③约57%国际多中心项目提供保险,而国内申办者普遍不为受试者购买保险,知情同意书上关于保险及赔偿问题,尤其对受试者如发生与试验相关的损害时,受试者可获得治疗和保险赔偿、经济赔偿等具体细节均未能详细列明。这些差异主要由于国外伦理审查较为完善,且对知情同意书有明确的监督管理机制和操作规程,而国内伦理审查仍处于逐步发展完善阶段,且国内药品临床试验管理规范没有明确规定统一的设计格式和标准,知情同意书各式各样。我院甚至有一项目的知情同意书分为两份,一份为受试者告知单,一份为受试者知情同意声明及双方签署部分,在归档资料中仅保留后者。这种做法是不规范的,知情同意书应是一份完整的文件。
在全球性的协作研究中,应学习国际科学的管理方法及研究设计等,逐步完善规范知情同意书的内容和格式,缩短与国际临床试验间的差距,切实保护受试者的权益。
3.2 知情同意书签字页设计可影响签署的完整性
本次调研发现,受试者的联系方式有43.70%因未设计而未签署,研究者的联系方式有17.29%因未设计而未签署。法定代理人代签时,有48.15%因未设计法定代理人与受试者的关系一栏而未签署。这就提示,可能部分未签署的现象源于签署设计项的缺失。因此,知情同意书的签字页设计全面,在一定程度上能规范受试者及研究者的签署行为。
3.3 知情同意书的签署存在不规范现象
本次调研中,知情同意书的签署存在不规范之处,主要表现为:①法定代理人代签时往往只填写代理人姓名,是不完整的,应同时填写受试者姓名及注明与受试者关系,以判断代理人是否符合代理人的条件[7]。②知情同意书中研究者的联系方式不填写或填写固定电话。在知情同意书中必须留下研究者的联系方式,且应填写手机号码,而非固定电话,否则受试者在试验期间出现不良事件时,就不能及时与研究者取得联系,给受试者的安全带来隐患。③知情同意书签署修改不规范。研究者自身应做到规范性修改,并能指导受试者进行规范性修改,且不得代受试者进行。
3.4 针对发现问题的对策
上述问题的发生,反映出我院对知情同意书的设计及审查体系需重视并改进,同时,也暴露出研究人员的职业素质需要强化。为了避免问题的重复发生,保证药物临床试验知情同意书的规范性,提出以下对策:①设计规范及标准操作规程的修订:机构办公室及专业科室对《知情同意书设计规范》和《知情同意的SOP》进行修订,进一步规范和统一知情同意书设计及签署的内容及流程。②加强教育与培训:加强对研究人员的职业道德教育,强化“以人为本”的服务理念。机构办公室及专业负责人组织研究者进行《药物临床试验质量管理规范》及知情同意专题的培训,要求其严格遵循“完全告知、充分理解、自主选择”的原则,规范性签署知情同意书。③提高伦理审查质量:伦理委员会应提高对知情同意书的审查标准及审查力度,切实保护受试者的权益,杜绝不规范的知情同意书。④健全机构内部检查制度,实施三级质控:本机构成立质量管理小组,实施机构办公室质控员、专业负责人、科室质控员的三级质量控制。提高对知情同意书的审查标准,定期对知情同意书的签署情况进行审查。发现问题及时反馈,提出改进措施,限期整改。本次调研中,未涉及知情同意的过程,以后的质控工作中,还应对知情同意的执行情况进行现场检查或向受试者进行电话咨询。
3.5 结语
知情同意书是临床试验成功与否的首要条件。在临床试验规范化、科学化建设的同时,做好知情同意真正体现以受试者为本的医学伦理、人道精神和行为[8]。通过本次调研,针对发现的问题提出对策,加以改进,实现对知情同意的规范化管理,只有这样才能使更多的患者从临床试验中受益,使我院的药物临床试验水平得到提高。
[1]国家食品药品监督管理局.药物临床试验质量管理规范[S]. 2003-09-01.
[2]王晓霞,李育民.我院药物临床试验实施知情同意工作的几点体会[J].中国药物与临床,2009,9(8):772-773.
[3]欧阳樱君.药物临床试验受试者知情同意的现状及改进措施[J].现代医院,2008,8(6):116-117.
[4]钟旋,刘秋生,刘大钺,等.药物临床试验知情同意书常见的伦理问题与对策[J].中国医学伦理学,2008,21(6):128-130.
[5]黄瑾,沈娜,刘厚佳,等.知情同意书信息要素完整性研究[J].药学服务与研究,2011,11(2):123-126.
[6]Mcguire DC,Chadwick GL.Protecting study volunteers in research[M].New York:Center Watch Inc/Thomson,2004:100.
[7]张蓉.知情同意现场检查中应关注的问题[J].中国医药导刊,2012,14(3):551-552.
[8]杜彦萍,杨忠奇,汪朝晖.对药物临床试验知情同意的解析[J].中医药管理杂志,2011,19(7):623-624.
Design and signature of informed consent form from clinical trial drugs of our hospital
GUO JinminZHANG LiSHU HeZHAO WenhuaKANG Changqing
Department of Pharmacy,Ji′nan Military General Hospital,Shandong Province,Ji′nan250031,China
ObjectiveTo discuss the design and signature of informed consent form(ICF)from clinical trial drugs of Ji′nan Military General Hospital(“our hospital”for short).MethodsItems of design and signature from clinical trial drugs were made,the occurrence ratio of every item in 2042 ICF from 58 clinical trial drugs in our hospital were analyzed, and items with lower occurrence ratio were explored.ResultsGenerally,the design and signature of ICF met the requirement of GCP.However,there were some defects of ICF and antonym of signature/revisions.In the domestic trail, the occurrence ratios of missing elements for design were lower than those in international trail,and the elements with low occurrence ratio were shown as follows:criteria of inclusion/exclusion(13.7%vs 71.4%),pay and compensation (7.8%vs 57.1%),the contact person and method of the Institutional Review Board(3.9%vs 85.7%),ICF version numbers and date(41.2%vs 100.0%).The elements for signature frequently missed were the contact information of subjects,contact information of investigators which fill in landlines or nothing,the name of subject and the relationship with subject which legal representative did not write.The absence of signature elements was further analyzed and found that it was partly due to the design defects.ConclusionStudies the causes and puts forward countermeasures to standardize informed consent,which promotes development of clinical trial in our hospital.
Clinical trial drugs;Informed consent form(ICF);Design;Signature
R197.3
B
1673-7210(2014)04(c)-0151-04
2013-11-19本文编辑:程铭)
郭晋敏(1983.2-),女,博士研究生;研究方向:心血管药理学和药物临床试验。
张莉(1972.12-),女,硕士研究生,副主任药师;研究方向:生化药学、药品质量控制、临床药学和药物临床试验。
猜你喜欢
保健与生活(2022年16期)2022-08-06
基层中医药(2020年5期)2020-09-11
上海医学(2019年8期)2019-02-12
小演奏家(2016年5期)2016-05-14
哈尔滨医药(2015年6期)2015-12-01
天津人大(2015年9期)2015-11-24
分忧(2014年9期)2014-09-22
哈尔滨医药(2014年5期)2014-02-27
中国合理用药探索(2012年2期)2012-03-20
中国合理用药探索(2011年9期)2011-03-20
