
新型掺杂多孔芳香骨架材料的储氢性能模拟
2014-02-18 12:07:02吴选军方继敏刘保顺蔡卫权
物理化学学报 2014年11期
吴选军 赵 鹏 方继敏 王 杰 刘保顺 蔡卫权,
(1武汉理工大学化学化工与生命科学学院,武汉430070;2武汉理工大学资源与环境工程学院,武汉430070;3武汉理工大学,硅酸盐建筑材料国家重点实验室,武汉430070)
1 引言
H2因具备来源广泛与清洁性等特点而成为未来车载燃料电池系统的主要能源之一,但现有压缩储存技术所能提供的储氢能量密度不到传统化石能源的一半.1为了提高车载储氢罐的储能密度和安全性,两种不同类型的储氢材料,即化学吸附储氢材料和物理吸附储氢材料受到研究人员的广泛关注.2-4前者主要是金属氢化物和复合氢化物等,如LiAlH4、LiBH4和氨硼烷等,5-7此类储氢材料的脱氢条件较苛刻,使其应用受到一定程度的限制.后者则包括金属有机骨架(MOFs)、8,9共价有机骨架(COFs)、10,11多孔芳香骨架(PAFs)12,13和多孔碳材料14,15(如碳纳米管、石墨烯和活性炭)等.这类储氢材料除了具备超高比表面积和超高孔隙率外,还具有良好的H2充放动力学特性,虽然较多类似多孔材料在冷冻温度下具有较高储氢能力,但是在常温下其储氢能力急剧下降,使得迄今为止还没有一种类似多孔材料在常温下能够达到美国能源部提出的2015年储氢目标,即在298 K、3.5-8.0 MPa下材料的重量储氢量达到5.5%(体积储氢量达到40 g∙L-1).1
PAFs材料由吉林大学朱广山与北京化工大学曹达鹏等共同设计、合成,是采用一个或多个苯环作为桥联体连接金刚石骨架中的碳原子而形成的一系列多孔骨架材料的总称.16,17其中PAF-1的BET比表面积达到5640 m2∙g-1,并且具有非常高的热稳定性和水热稳定性.16为了提高PAFs材料的常温储氢能力,研究人员通过对材料中苯环结构进行功能化修饰或金属掺杂,18提高吸附剂分子与客体分子之间的相互作用能,以达到增大材料储氢量的目的.Cao等19,20通过理论预测了PAF-304在常温、10.13 MPa下H2吸附量达到6.53%(w),Sun等21则采用四氮唑锂盐修饰PAF-303材料中的苯环,并预测其在233 K、10 MPa下的H2吸附量达到4.9%(w).Jiang等22在PAFs材料的联苯单元上引入两亲性有机基团类四氢呋喃醚,巨正则蒙特卡洛模拟(GCMC)模拟结果表明,改性后的PAFs材料在常温常压下的 CO2吸附量达到 10 mmol∙g-1,对 CO2/H2、CO2/CH4和CO2/N2等混合物体系表现出较高的CO2吸附选择性.Ahmed等23通过分子设计将锂修饰富勒烯(LiC60)植入PAFs材料孔道中,分子模拟结果表明,植入LiC60后的PAFs材料储氢能力大幅提高,在1 MPa、77 K下的体积储氢量从12 g∙L-1提高到44 g∙L-1,增幅为260%.锂掺杂也被应用到其它类似多孔材料中,如Yang和Cao11研究了锂掺杂对COFs材料中H2和CH4的扩散与分离性质的影响规律,发现锂掺杂后的COFs材料对CH4/H2混合物体系的吸附选择性大幅度增加.Cao等20报导了锂掺杂COF-105和COF-108的重量储氢能力,GCMC模拟结果显示,在298 K、10 MPa下它们的H2吸附量分别达到6.84%(w)和6.73%(w).
由于苯环具有芳香性,导致掺杂Li等金属原子与苯环之间的相互作用力较弱,甚至低于金属Li中原子内能(163 kJ∙mol-1),因此掺杂的Li原子存在聚集成簇的趋势.1Srinivasu和Ghosh24报导了在苯环中引入B原子可以促进Li原子离子化,从而提高Li原子与苯环之间的相互作用力,理论计算表明每个Li原子与C4B2H6之间的结合能为-359.2 kJ∙mol-1,由C4B2H6Li2单元组成的改性MOF-5每个分子单元能够吸附18个H2,相当于重量储氢量约为4.3%(w).虽然理论预测能够获得较高储氢能力,但没有得到进一步的证实.
本文借鉴了Srinivasu和Ghosh24报导的Li掺杂方法,采用C4B2H6Li2单元取代PAF-301材料中的苯环结构,得到PAF-C4B2H4-Li2骨架材料,为了与简单的Li掺杂效果进行比较,也构建了PAF-301Li的模型;同时参考王维25和元野26等设计以Si和Ge为骨架中心构建PAFs新材料的思路,将PAF-C4B2H4-Li2骨架中心C原子分别用Si和Ge原子取代,又得到PAF-C4B2H4-Li2-Si和PAF-C4B2H4-Li2-Ge两种PAFs新材料.为了研究它们的储氢性能与其结构之间的关系,本文选用多尺度计算方法对PAFs材料进行理论研究,首先从不同PAFs材料中选取一定的分子片段,利用密度泛函理论(DFT)和Moller-Plesset二阶微扰理论(MP2)模型获得分子片段与H2分子间的结合能,同时利用DDEC(density-derived electrostatic and chemical charge method)方法27计算以上各分子片段的电荷分布,以研究各分子片段与H2分子间的相互作用规律.然后采用量子力学方法对各种PAFs材料进行构型优化,并在最优构型基础上通过GCMC方法计算材料在常温和低温下的储氢性能,通过讨论77 K下H2在各种PAFs材料中达到吸附平衡时的质心密度分布和等位能面分布特征,对引入锂、硼等原子掺杂模式影响PAFs材料储氢性能的机理进行了初步分析.
2 分子模型与理论计算
2.1 分子模型与力场参数
2.1.1 分子模型建立
PAF-301的晶体模型根据Ben等28报导P1模型结构建立,其单元晶胞分子式为C104H64(见图1(a)),单元晶胞大小为1.37651 nm×1.37651 nm×1.37651 nm的立方型结构,骨架中心C原子形成类似金刚石拓扑结构.其它几种改性的PAFs材料骨架结构均在PAF-301基础上建立,其中骨架中心C原子被Si或Ge原子取代,同时苯环上的两个对位C原子被B原子替代,并在每个六元环结构中掺杂进1或2个Li原子,形成的骨架结构单元晶胞大小与PAF-301相同,单元晶胞分子式及比表面积等性质在表1中列出,单元晶胞分子结构式以及三维晶胞构型见图1.
2.1.2 力场参数
在GCMC模拟过程中,PAFs材料采用刚性骨架,即骨架中各原子位置保持固定,只考虑H2分子之间和H2与PAFs骨架原子之间的相互作用.29H2与PAFs材料之间的相互作用属于弱相互作用,且H2分子极化程度较小,故只须考虑Lennard-Jones(LJ)位能项,即:
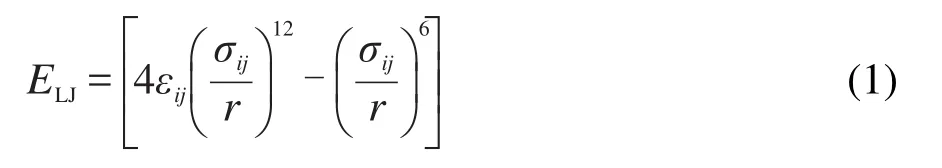
式中,r为第i和第j个原子之间的距离,σij为碰撞直径,εij为能量参数,不同原子之间的交叉作用参数εij和σij采用Lorentz-Bertelot混合规则计算:30,31

H2与PAFs骨架原子之间LJ相互作用参数的选取非常重要,直接决定GCMC模拟结果的好坏.鉴于采用通用力场(UFF)和DREIDING力场均在预测MOFs和COFs等材料中小分子气体的等温吸附曲线时取得较大成功,32,33本文选用以上两种力场中的LJ相互作用参数来描述H2与PAFs骨架原子之间的相互作用,各原子的力场参数见表2.由于DREIDING力场中缺少Li原子的LJ相互作用参数,故在采用DREIDING力场计算时Li原子的LJ相互作用参数选自UFF力场.34
2.2 理论计算方法
2.2.1 量子力学计算方法
对于非周期性分子片段,分别根据DFT和MP2理论采用GAMESS软件35进行构型优化与能量计算.所选非周期性分子片段为构成PAFs材料的几种有机桥联体,分别为C6H6Li和C4B2H6Li2等体系(见表3).DFT计算选用混合泛函B3LYP,与MP2计算一样均采用6-31++G(2d,2p)扩展劈裂价电基组带扩散与极化函数描述体系波函数.由于分子片段与H2之间的相互作用较弱,在计算分子片段与H2分子结合能时通过CounterPoise校正方法36来系统消除复合物体系的基组重叠误差(BSSE),分子片段与H2分子结合能(EB)按下式计算:
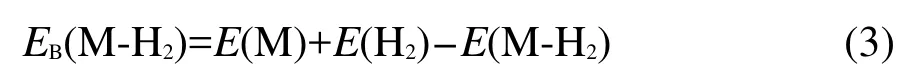
本文首先通过以上两种不同的方法对不同非周期性分子片段进行了构型优化,然后在优化后的构型基础上计算了不同分子片段与单个H2分子之间的结合能,同时利用DDEC方法分别计算了分子片段单独存在和与单个H2分子结合时的原子电荷分布.对于周期性的PAFs晶体结构,则采用量子力学计算方法进行初步构型优化,优化过程由VASP软件完成,37采用平面缀加波(PAW)基组和PBE赝势,所有电子扩充缀加波函数截断为400 eV,因为处理的晶胞较大,运用Monkhorst-Pack方法产生的1×1×1的k网格对布里渊区域进行积分,所有晶体中的原子位置完全松弛没有任何约束.
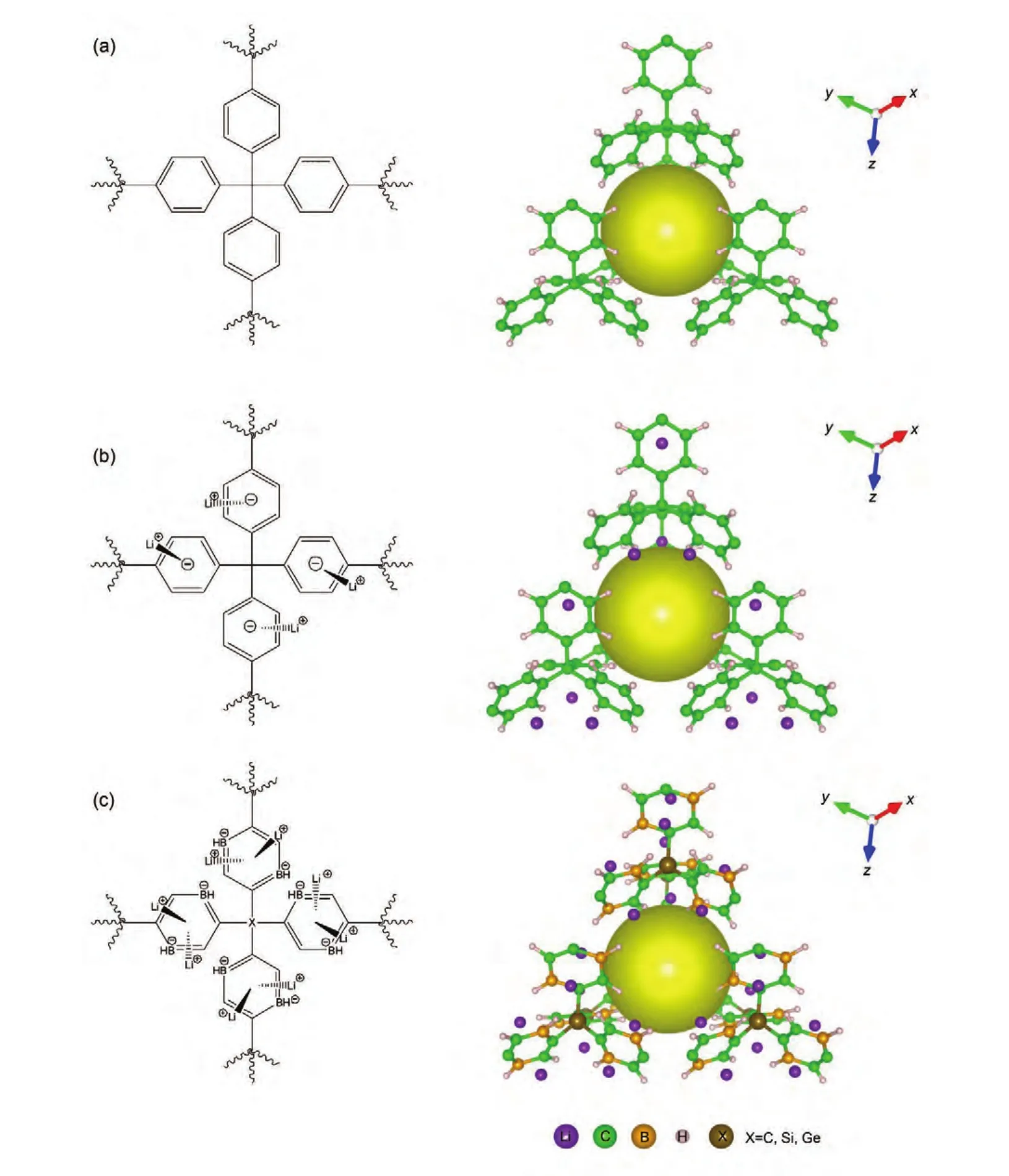
图1 不同PAFs材料的分子结构式与单元晶胞三维结构Fig.1 Molecular structures and three-dimensional unit cell structures of various PAFs
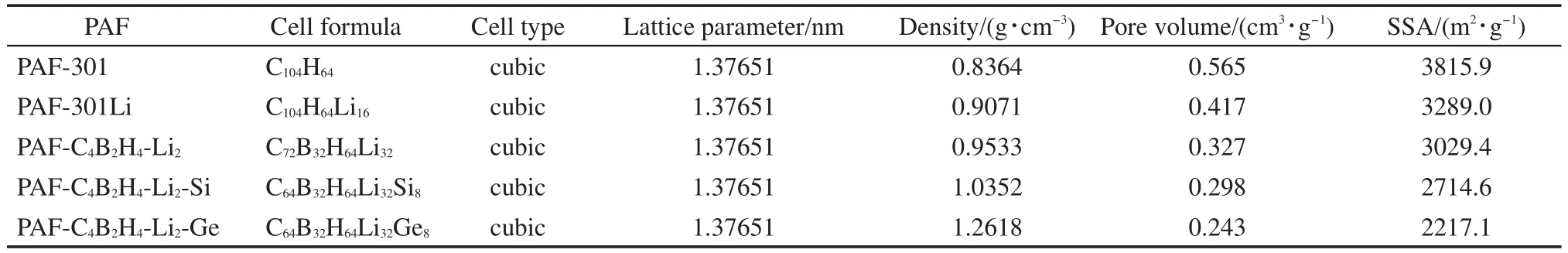
表1 不同PAFs材料的分子式及各种性质Table 1 Formula and various properties of different PAFs
2.2.2 GCMC模拟
采用GCMC方法模拟H2分子在不同PAFs材料中的吸附等温曲线,模拟程序为Snurr研究组38提供的MUSIC-4.0.模拟盒子包含2×2×2的超级晶胞,三个方向上均设为周期性以消除边界效应.H2分子采用全原子模型,39被当作球形分子,其LJ相互作用参数分别为ε/kB=36.7 K,σ=0.2958 nm,29其中kB为玻尔兹曼常数.由于H2分子不带电荷,所以关闭原子之间的静电相互作用,LJ相互作用的截断半径设为1.2 nm,GCMC模拟总循环步数为107步,前5×106步为平衡时间,后5×106步为采样时间,每隔1000步输出一次构型,用于质心密度分布统计计算.GCMC方法统计的H2分子吸附量有两种,分别为过量吸附量与绝对吸附量,两者之间的关系可以通过下式转换:40
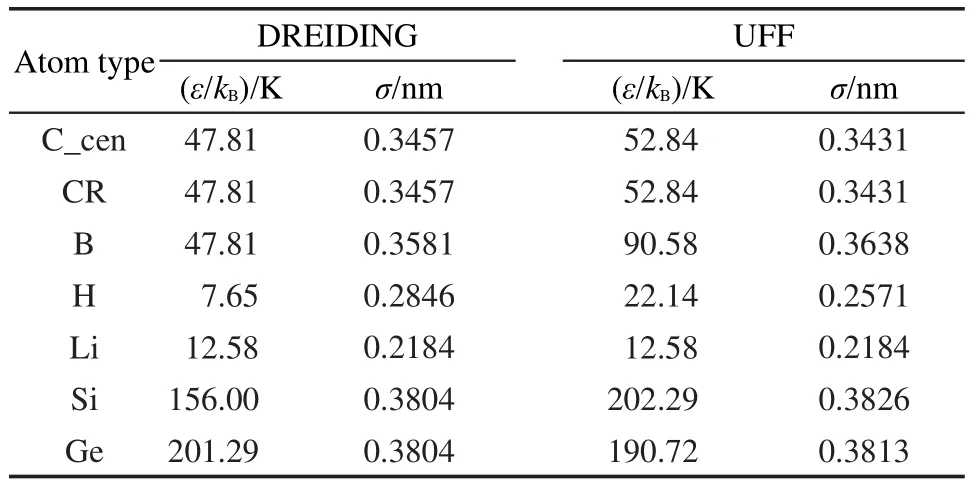
表2 PAFs材料骨架中各原子的力场参数Table 2 Force field parameters for all atoms in frameworks of PAFs

式中,Nexc和Nabs分别为过量吸附量与绝对吸附量,ρbulk和Vfree分别为吸附质H2分子在主体相中的密度和吸附骨架的自由体积,其中ρbulk采用Peng-Robinson状态方程41根据温度和压力计算得到,Vfree则以N2分子为探针通过Duren等42提供的方法获取(见表1).
3 结果与讨论
3.1 分子片段与H2分子结合能及电荷分布
3.1.1 分子片段构型优化及与H2分子间的结合能
由于PAFs晶胞较大,从PAFs材料骨架中选取了几种不同的分子片段(见表3)进行量子力学计算,以减少计算资源消耗.先通过第一性原理对各分子片段进行构型优化,优化后的分子片段结构见图2.当单个Li原子与苯环相互作用时,Li-C间距为0.236 nm,但当两个Li原子与苯环相互作用时,Li-C间距增加到0.271 nm,说明引入更多的Li原子后Li与苯环的相互作用减弱.如果将苯环中的两个C原子替换为B原子,由于B原子核比C原子核小,对核外价电子的约束能力也较后者低,从而会失去部分电子带上正电荷,但对整体而言会形成一个带负电的六元环能够与两个Li原子形成更稳定的结构,C4B2H6-Li2优化后的Li-C间距和Li-B间距分别缩小至0.219-0.221 nm和0.225-0.227 nm.
Srinivasu和Ghosh24采用MP2/6-31++G(2d,2p)方法分别计算了C4B2H6-Li2和C6H6-Li2中Li原子与中性六元环之间的相互作用能,结果显示每个Li原子与C4B2H6之间的相互作用能为-353.9 kJ∙mol-1,而与C6H6之间的相互作用能仅为-32.13 kJ∙mol-1,进一步证实C4B2H6-Li2结构更加稳定.本文进一步对吸附一个H2分子的分子片段体系进行构型优化,发现与 C6H6-Li、C6H6-Li2体系相比,在 C4B2H6-Li2与 H2相互作用的体系中,Li-H2间距更大一些,从0.207-0.205 nm增加到0.217 nm,与Srinivasu和Ghosh计算24的Li-H2相当(约0.220 nm).H2与不同分子片段之间的结合能计算采用了BSSE修正,计算结果见表3.在没有掺杂Li之前,H2与六元环C6H6和C4B2H6之间的结合较弱,采用B3LYP/6-31++G(2d,2p)方法计算的结果甚至表明两者互相排斥;掺杂一个Li原子后,由于Li原子带正电荷,对H2分子有较强的吸引力,H2与C6H6-Li和C4B2H6-Li之间结合能明显提高,尤其是H2与C4B2H6-Li之间结合能达到-16.07--17.20 kJ∙mol-1;掺杂两个Li原子后,H2与C6H6-Li2和C4B2H6-Li2之间的结合能进一步提高.
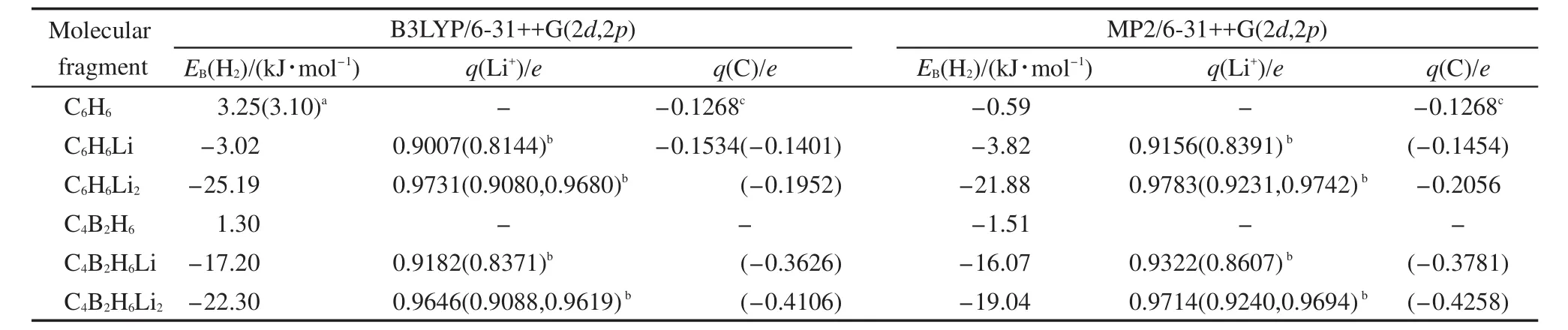
表3 PAFs分子片段与H2的结合能及碳原子、锂离子电荷Table 3 Binding energy between H2and PAFs fragments,partial charges of carbon atoms and Li ions
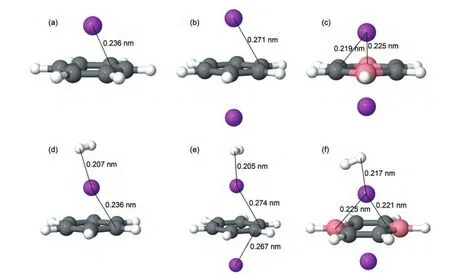
图2 优化后各分子片段的结构Fig.2 Optimized structures of various molecular fragments
3.1.2 分子片段电荷分布
在采用GCMC或分子动力学计算极性流体在MOFs等多孔材料中的吸附与扩散性质时,必须考虑材料骨架中各原子所带电荷,原子部分电荷值往往通过量子力学方法计算.但迄今为止,尚无关于PAFs材料原子部分电荷的文献报导.与传统的Mulliken布居分析法和静电势拟合法相比,DDEC采用同时拟合材料化学态和电子密度分布静电势的方法,避免了原子电荷计算的基组依赖性,并且具有较强的可移植性.27采用量子化学方法直接对三维晶体结构进行原子电荷计算非常占用计算资源,本文选用DDEC方法分别基于B3LYP/6-31++G(2d,2p)和MP2/6-31++G(2d,2p)对各分子片段中原子部分电荷进行计算(Li、C原子电荷见表3),发现基于 B3LYP/6-31++G(2d,2p)和MP2/6-31++G(2d,2p)计算的结果相差不大,说明DDEC方法对基组的依赖性较弱.计算所得各分子片段中Li原子的电荷均在+0.8e以上,表明Li原子掺杂到C6H6和C4B2H6中后均表现出较强的离子特性,掺杂Li以后的分子片段再与H2相互作用时,Li原子电荷均出现不同程度的下降,其中与H2分子处于六元环平面同一侧的Li原子电荷下降明显,而处于异侧的Li原子电荷只出现轻微下降,这是由于Li离子与之相邻的H2分子之间产生了明显的电荷转移,即对H2分子形成极化效应.两种六元环C6H6和C4B2H6分子结构中掺杂两个Li原子比掺杂一个Li原子时Li原子的电荷更高,在相同掺杂情况下,C6H6和C4B2H6分子片段中Li原子电荷只有较小差别,掺杂一个Li原子时,前者Li原子电荷较小,而掺杂两个Li原子时情况则相反.
在六元环中引入B原子后,其中C原子的电负性表现得更加明显,而B原子则带正电荷.分子片段在掺杂Li原子后,C原子的电负性进一步增强,掺杂的Li原子数量越多,C原子的电负性越强;但与H2分子相互作用后,C原子的电负性出现适度下降.
3.2 H2在不同PAFs材料中的吸附特性
3.2.1 77 K下H2在PAFs材料的吸附等温曲线
77 K下H2在MOFs等多孔材料中的吸附往往表现出量子效应,为了让模型更简单,本文选择了Frost和Snurr29报导的H2全原子模型,该模型在忽略Feynman-Hibbs量子效应时也能很好地描述77 K下H2在MOFs材料中的吸附等温线.29
图3为采用DREIDING力场描述PAFs材料不同骨架原子时由GCMC模拟得到的77 K下H2在PAFs材料中的吸附等温线,其中图3(a)和图3(b)分别代表以单位质量和单位体积吸附剂为基准的H2过量吸附量(下同),以表征PAFs材料的质量吸附能力和体积吸附能力.由图中曲线可以看出,与PAF-301相比,PAF-301Li骨架由于掺杂Li原子后晶体密度增加,单位质量的自由孔体积有所下降,但在高压下其对H2的吸附量并没有因为自由孔体积减小而出现下降,反而超过PAF-301对H2的吸附量,表现出较好的增强效果,其饱和质量吸附量超过5.0%(w),超过文献43报导78 K下MOF-5的储氢量(4.5%(w)),但与其它储氢性能强的多孔材料相比,PAF-301Li骨架的储氢性能还有待进一步提高(见表4);从体积吸附量上来看,PAF-301Li对H2的吸附能力也是最强的,最大体积吸附量(标准状态,STP)超过500 cm3∙cm-3.PAF-C4B2H4-Li2骨架的H2质量吸附量与H2体积吸附量均低于PAF-301,尤其是二者H2质量吸附量存在明显差距.这归因于掺杂Li原子后PAF-C4B2H4-Li2骨架的单位质量自由孔体积出现明显下降,由0.565 cm3∙g-1下降到0.327 cm3∙g-1,单位质量自由孔体积的下降使其对H2的质量吸附量降低.二者在高压下的H2体积吸附量非常接近,根据前面计算的结合能数据,由于PAF-C4B2H4-Li2骨架原子对H2具有更强的吸附能力,使其在高压下保持了相对高的H2体积吸附量.由于Si和Ge的原子量相对较大,使得PAF-C4B2H4-Li2-Si和PAF-C4B2H4-Li2-Ge骨架单位质量自由孔体积下降到只有0.298和0.243 cm3∙g-1,从而导致它们的H2质量吸附能力和H2体积吸附能力较PAF-301更低,并且二者的H2体积吸附能力几乎保持一致,这是因为骨架中心Si和Ge原子与H2的相互作用能差别不大,而且骨架中心原子数量也比较少的缘故.

图3 DREIDING力场模拟77 K下H2在PAFs材料中的吸附等温线Fig.3 Adsorption isotherms of H2on PAFs at 77 K simulated by DREIDING force field
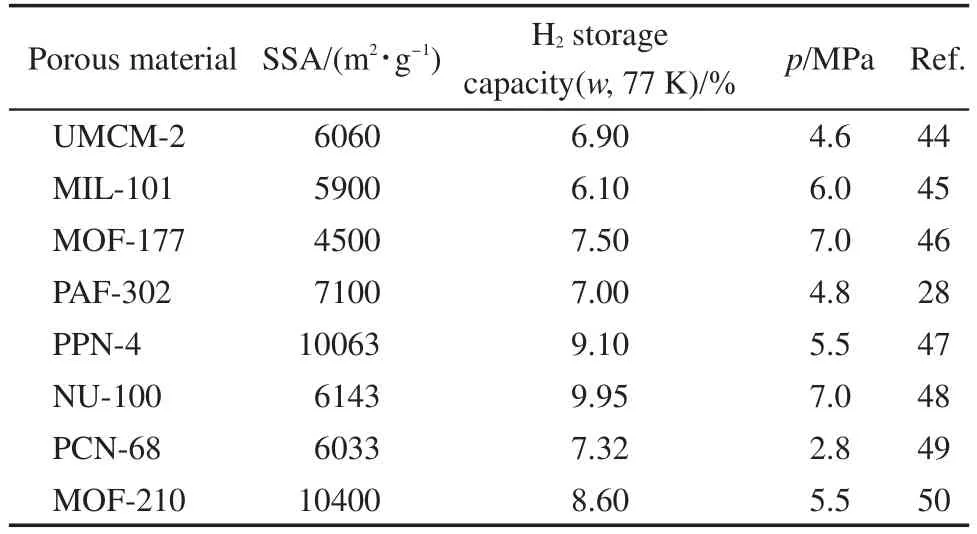
表4 不同多孔骨架材料的比表面积与储氢性能Table 4 Specific surface areas and H2storage capacities of different porous frameworks
图4为UFF力场模拟77 K下H2在PAFs材料中的吸附等温线.图4与图3的区别在于对不同PAFs材料骨架原子采用UFF力场进行GCMC模拟得到77 K下H2在PAFs材料中的吸附等温线.在UFF力场中,B原子的能量参数远高于DREIDING中B原子的能量参数,其它原子的力场参数也与DREIDING中相应原子的力场参数有一定差别(除Li原子外),使得图4中77 K下H2在不同PAFs材料中的吸附等温线规律与图3存在较大差异性.从图4可知,PAF-301Li依然表现出较好的储氢能力,高压下其H2质量吸附量和体积吸附量最高,而低压下H2吸附量较小,这将有利于H2的释放.相对PAF-301和PAF-301Li而言,PAF-C4B2H4-Li2、PAF-C4B2H4-Li2-Si和PAF-C4B2H4-Li2-Ge三种骨架分别采用UFF力场与DREIDING力场预测的储氢能力相差较大,尤其是采用UFF力场预测的PAF-C4B2H4-Li2-Si和PAFC4B2H4-Li2-Ge高压下体积储氢能力超过PAF-301,主要归因于两种力场对骨架材料中B、Si和Ge原子的描述存在较大差异,力场参数存在进一步优化的空间.
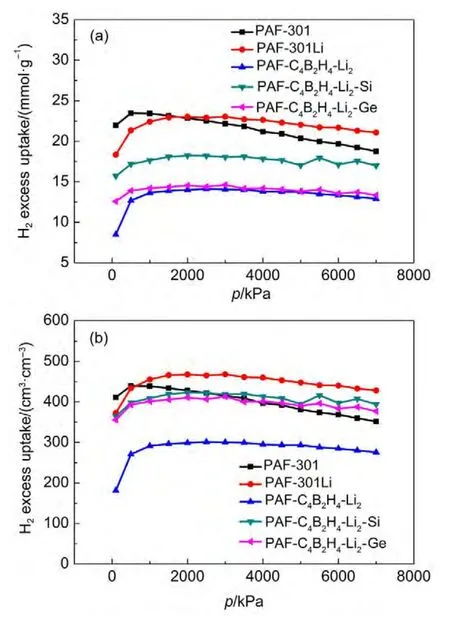
图4 UFF力场模拟77 K下H2在PAFs材料中的吸附等温线Fig.4 Adsorption isotherms of H2on PAFs at 77 K simulated by UFF force field
3.2.2 298 K下H2在PAFs材料的吸附等温曲线
以H2为能源的燃料电池工作温度为-40-85°C,工作压为0.15-10.00 MPa,研究常温下PAFs材料对H2的吸附规律对开发燃料电池储氢材料具有重要现实意义.本文分别采用DREIDING力场和UFF力场对298 K下H2在不同PAFs材料中的等温吸附曲线进行了GCMC模拟,结果分别见图5和图6,图中也分别列出了质量吸附量和体积吸附量随着压力增大而产生的变化.
从图5可知,采用DREIDING力场模拟的结果表明PAF-301骨架材料具有最佳的常温储氢性能,不过与77 K下的等温吸附曲线相比较,常温下不同PAFs材料的最大储氢能力均出现大幅下降,如PAF-301骨架材料的最大储氢能力从77 K下的5.0%(w)下降到298 K时的约0.7%(w).PAF-C4B2H4-Li2骨架材料表现出了较好的常温储氢性能,质量储氢量超过了PAF-301Li而仅次于PAF-301,体积储氢量与PAF-301几乎相等,根据前面量子化学计算的结果,这主要归因于PAF-C4B2H4-Li2在常温下与H2分子之间存在较强相互作用.PAF-C4B2H4-Li2-Si和PAFC4B2H4-Li2-Ge骨架材料的常温储氢性能相似,吸附等温线几乎重合.
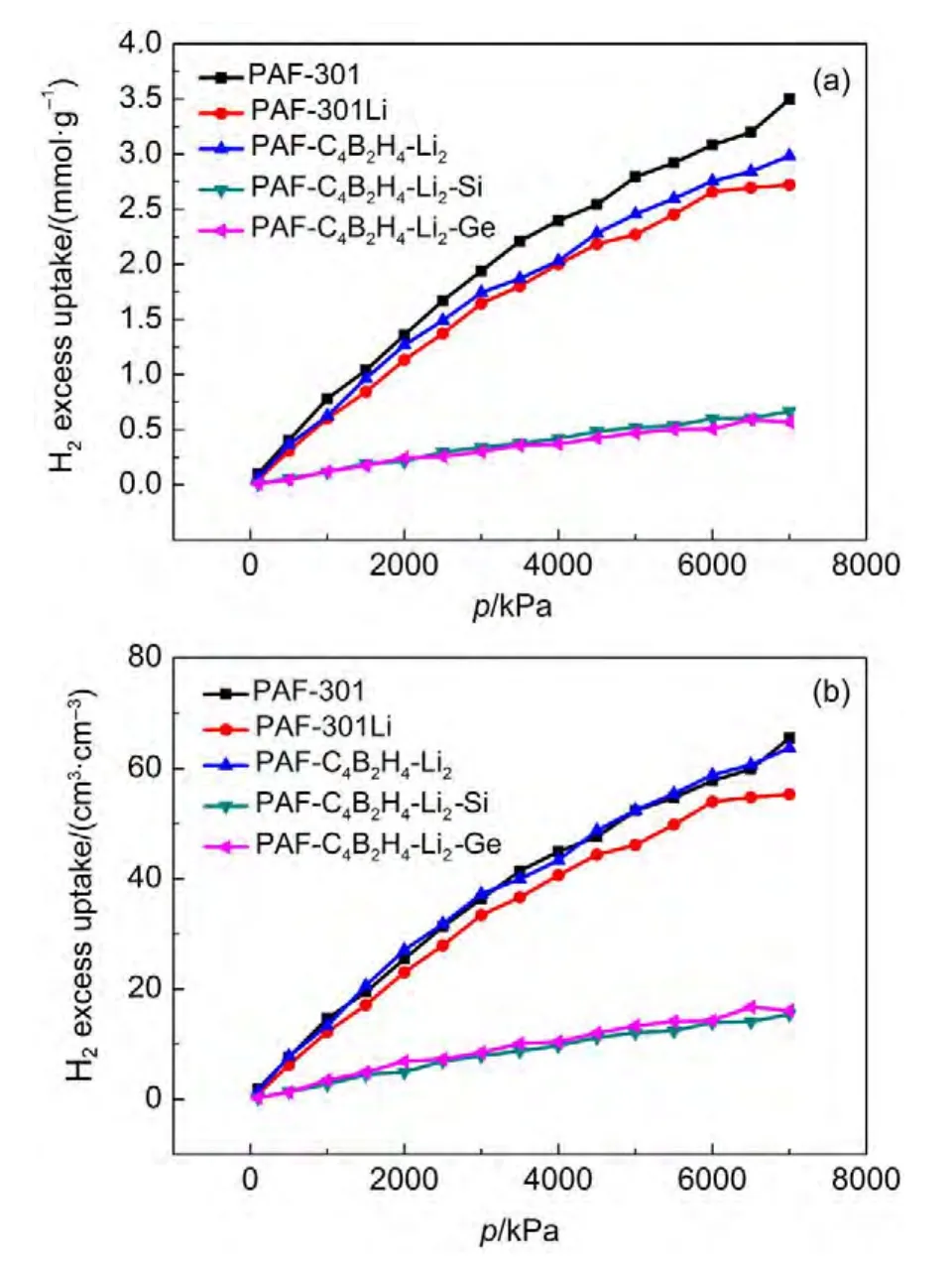
图5 DREIDING力场模拟298 K下H2在PAFs材料中的吸附等温线Fig.5 Adsorption isotherms of H2on PAFs at 298 K simulated by DREIDING force field
从图6可知,常温下H2质量吸附量依次递减的顺序是PAF-C4B2H4-Li2-Si>PAF-C4B2H4-Li2-Ge>PAF-301>PAF-301Li>PAF-C4B2H4-Li2,常温下H2体积吸附量也表现出类似规律,不过PAF-301与PAF-301Li、PAF-C4B2H4-Li2-Si与PAF-C4B2H4-Li2-Ge两组骨架材料的常温H2体积吸附量曲线几乎重合.
3.3 H2在不同PAFs材料中的优先吸附位置
3.3.1 77 K下H2在PAFs材料中的等位能面
为了明确H2在不同PAFs材料中的优先吸附位置,本文首先以H2作为探针分子计算了各种PAFs材料单元晶胞内部的等位能面分布(见图7).对于PAF-301晶胞,普遍的等位能面处于较低取值范围,只有零星的位置点位能超过1046 kJ∙mol-1,引入Li原子后,局部等位能面取值明显增加,说明掺杂Li原子后使得整个体系能量提高,增加了体系的不稳定性,但是能量较低的等位能区域分布特征变化不大.随着骨架苯环中B原子的引入,能量较低的等位能区域的分布形态发生明显变化,中心部分整个区域的宽度被显著压缩,而这些区域正是H2在PAFs材料中的优先吸附位置.

图6 UFF力场模拟298 K下H2在PAFs材料中的吸附等温线Fig.6 Adsorption isotherms of H2on PAFs at 298 K simulated by UFF force field
在PAF-C4B2H4-Li2-Si和PAF-C4B2H4-Li2-Ge骨架单元晶胞中,能量较低的等位能区域被进一步压缩在更小的范围之内,但其分布形态没有再发生进一步的变化.
3.3.2 77 K下H2在PAFs材料的吸附平衡质心密度分布
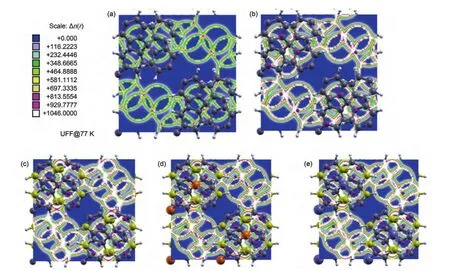
图7 77 K下H2在PAFs材料中吸附的LJ等位能面(xoy)Fig.7 LJ iso-potential surface(xoy)for H2on PAFs at 77 K
为了进一步明确H2在不同PAFs材料中的优先吸附位置,对77 K下H2在PAFs材料中达到吸附平衡时的构象进行了统计处理,并利用晶体可视化软件Xcrysden51得到了H2在PAFs材料中的吸附平衡质心密度分布图.对每一种PAFs骨架晶体,从GCMC计算中的500-1000万步进行取样,每隔1000步输出一次构型,然后对所有构型进行质心分布统计,即将单元晶胞分成100×100×100格子,统计H2在各个格子出现的概率,并进行归一化处理,即在整个单元晶胞内出现的概率为1;对每一种PAFs骨架晶体分别统计了三种不同压力下的吸附平衡数据.
图8和图9分别为PAF-301Li和PAF-C4B2H4-Li2中H2达到吸附平衡时的质心分布图,其它三种骨架晶体PAF-301、PAF-C4B2H4-Li2-Si和PAF-C4B2H4-Li2-Ge中H2达到吸附平衡时的质心分布图见图S1-S3(Supporting Information).
77 K下H2在PAF-301和PAF-301Li骨架中吸附平衡质心密度分布图显示这两种晶胞都体现出一定的对称性,这与H2在两种材料中的等位能面分布形态相一致,由于晶胞中心能量较低的等位能区域较宽,出现了四个明显的H2分子高密度分布区域;但Li原子的引入使得H2在后者骨架中的优先吸附位置出现一定程度偏移,H2在PAF-301Li骨架中的优先吸附位点趋向更加集中,其中两个H2分子高密度分布区域得到增强、扩大,而在另一交叉方向上的两个较高H2分子质心密度区域出现弱化.从77 K下H2在PAF-301Li骨架中的吸附等温曲线来看,PAF-301Li具有更高的储氢性能,因而H2分子高密度分布区域得到增强、扩大,但这种增强与扩大具有很强的区域性和方向性.从不同压力下的H2吸附平衡质心密度分布来看,压力对质心密度分布形态的影响不大,但是随着压力的提高,H2的优先吸附位点更加集中,3.5和7.0 MPa下的质心密度分布相差不大,说明随着压力的逐渐提高,H2在PAFs材料中的吸附逐渐达到饱和,H2的优先吸附位置也不再出现明显变化,这与吸附等温曲线的变化趋势保持一致.
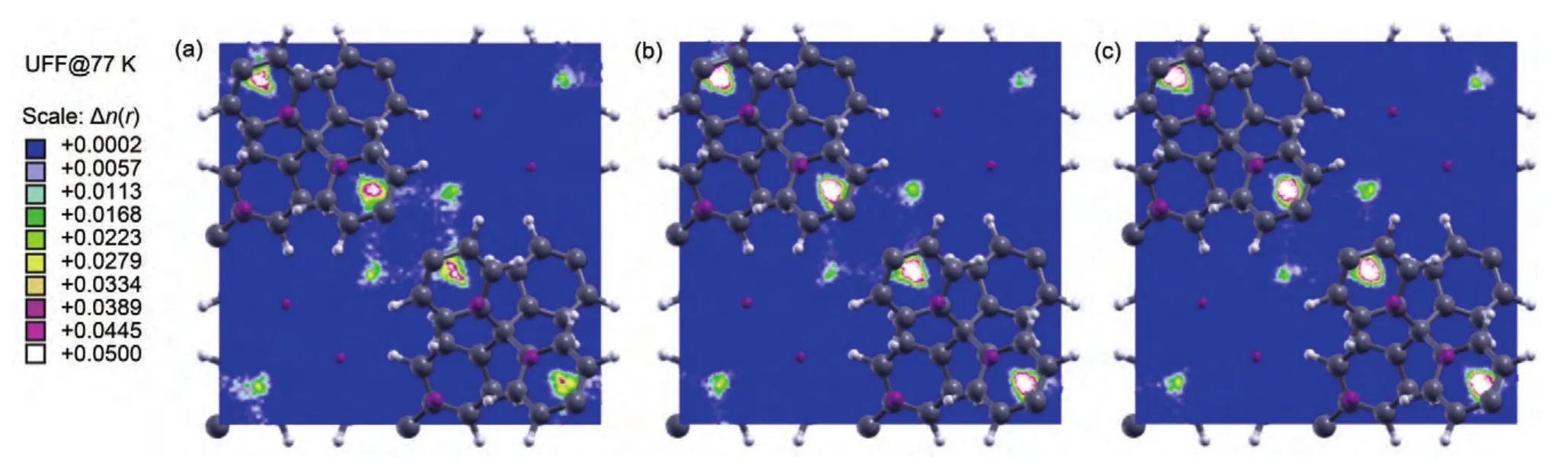
图8 77 K不同压力下H2在PAF-301Li中吸附平衡质心密度分布(xoy)Fig.8 Mass center density distribution(xoy)for adsorption equilibrium of H2on PAF-301Li at various pressures and 77 K
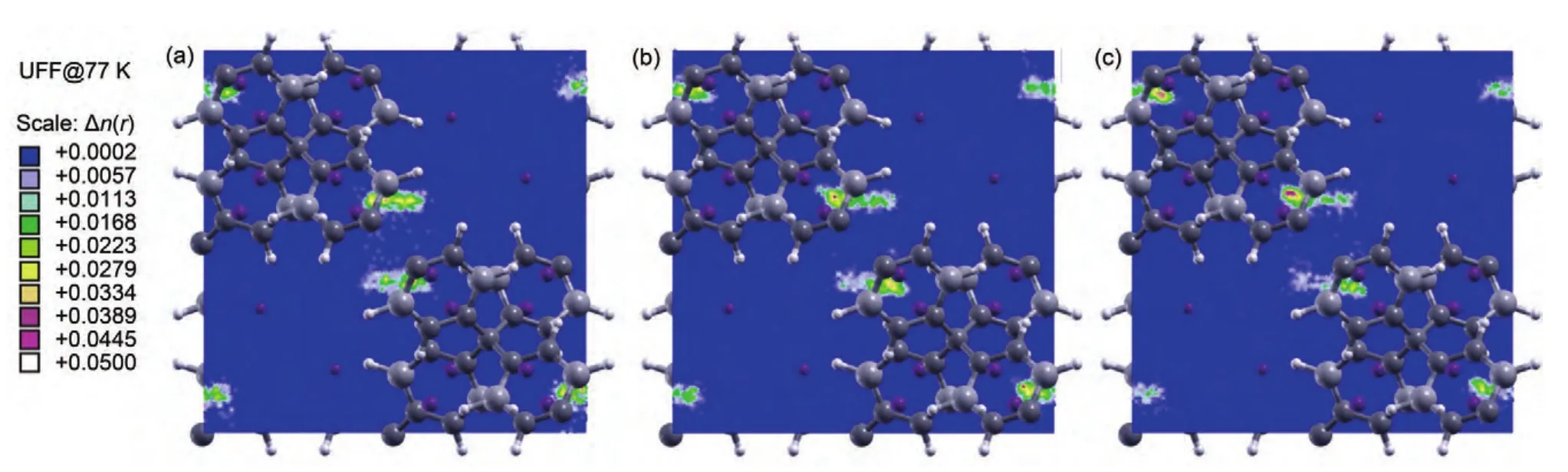
图9 77 K不同压力下H2在PAF-C4B2H4-Li2中吸附平衡质心密度分布(xoy)Fig.9 Mass center density distribution(xoy)for adsorption equilibrium of H2on PAF-C4B2H4-Li2at various pressures and 77 K
77 K下H2在PAF-C4B2H4-Li2、PAF-C4B2H4-Li2-Si和PAF-C4B2H4-Li2-Ge骨架中吸附平衡质心密度分布形态与前两者明显不同,晶胞中心只出现两个明显的H2分子高密度分布区,结合77 K下H2在PAFs材料中的等位能面分布形态推测,由于在这三种晶体结构中,中心能量较低的等位能区域压缩至很小的范围,使得在前面两种骨架晶体PAF-301和PAF-301Li中心出现的四个H2分子高密度分布区产生相互重叠而形成两个新的H2分子高密度分布区.在PAF-C4B2H4-Li2晶胞中,不论什么压力下H2分子高密度分布区的强度和范围均不大,这正好应证了其具有较低的储氢性能.
4 结论
基于PAF-301骨架材料通过Li原子掺杂或B原子取代等模式设计了几种新型PAFs材料,采用量子力学和分子力学方法对新材料储氢性能的影响因素进行研究.利用DFT理论和MP2理论对不同分子片段进行构型优化,并计算了各分子片段与H2之间的结合能,采用DDEC方法计算了各分子片段中原子电荷分布.分别选择DREIDING力场和UFF力场参数利用GCMC模拟方法计算了77和298 K下H2在不同PAFs材料中的吸附平衡性质.结果表明,H2直接与苯环的结合能较低,但掺杂Li原子能够提高H2与六元环的结合能,同时Li原子体现出较高的正电性质,B原子取代苯环中的两个C原子后,使得原有C原子电负性增强;77 K下PAF-301Li具有最高的储氢性能,而PAF-C4B2H4-Li2-Si和PAF-C4B2H4-Li2-Ge体现出较好的常温储氢性能,各种骨架材料的常温储氢性能远低于其低温储氢性能.通过77 K下H2在PAFs材料中的等位能面分布和吸附平衡质心密度分布对H2在PAFs材料中的优先吸附位置进行分析,发现在PAF-301和PAF-301Li骨架中,由于中心能量较低的等位能区域范围较宽,H2在其中存在四个明显的吸附高密度分布区域,而其它三种PAFs晶胞中心能量较低的等位能区域范围较窄,使得H2在其中只存在两个明显的吸附高密度分布区域.研究表明,B原子引入到苯环中并未如文献预期那样提高PAF-C4B2H4-Li2的储氢性能,但意外预测到PAF-C4B2H4-Li2-Si和PAF-C4B2H4-Li2-Ge具备较高的常温储氢性能,不过还有待进一步理论或实验验证.
Supporting Information:available free of chargeviathe internet at http://www.whxb.pku.edu.cn.
(1) Furukawa,H.;Cordova,K.E.;O'Keeffe,M.;Yaghi,O.M.Science2013,341(6149),974.
(2) Han,S.S.;Jung,D.H.;Choi,S.H.;Heo,J.ChemPhysChem2013,14(12),2698.doi:10.1002/cphc.v14.12
(3)Guo,J.H.;Zhang,H.;Miyamoto,Y.Phys.Chem.Chem.Phys.2013,15(21),8199.doi:10.1039/c3cp50492a
(4)Yang,Q.;Liu,D.;Zhong,C.;Li,J.R.Chem.Rev.2013,113(10),8261.doi:10.1021/cr400005f
(5) Pathak,B.;Pradhan,K.;Hussain,T.;Ahuja,R.;Jena,P.ChemPhysChem2012,13(1),300.doi:10.1002/cphc.201100585
(6)Varin,R.A.;Zbroniec,L.J.Alloy.Compd.2010,506(2),928.doi:10.1016/j.jallcom.2010.07.119
(7)Xu,J.;Qi,Z.;Cao,J.;Meng,R.;Gu,X.;Wang,W.;Chen,Z.Dalton Trans.2013,42(36),12926.doi:10.1039/c3dt50933h
(8) Tranchemontagne,D.J.;Park,K.S.;Furukawa,H.;Eckert,J.;Knobler,C.B.;Yaghi,O.M.J.Phys.Chem.C2012,116(24),13143.doi:10.1021/jp302356q
(9)Zhou,H.C.;Long,J.R.;Yaghi,O.M.Chem.Rev.2012,112(2),673.doi:10.1021/cr300014x
(10) Mendoza-Cortes,J.L.;Goddard,W.A.,III;Furukawa,H.;Yaghi,O.M.J.Phys.Chem.Lett.2012,3(18),2671.doi:10.1021/jz301000m
(11) Yang,Z.;Cao,D.J.Phys.Chem.C2012,116(23),12591.doi:10.1021/jp302175d
(12) Ben,T.;Qiu,S.CrystEngComm2013,15(1),17.doi:10.1039/c2ce25409c
(13)Miao,Y.L.;Sun,H.;Wang,L.;Sun,Y.X.Acta Phys.-Chim.Sin.2012,28(3),547.[苗延霖,孙 淮,王 琳,孙迎新.物理化学学报,2012,28(3),547.]doi:10.3866/PKU.WHXB201112301
(14) Hussain,T.;De Sarkar,A.;Ahuja,R.Int.J.Hydrog.Energy2014,39(6),2560.doi:10.1016/j.ijhydene.2013.11.083
(15) Gopalsamy,K.;Subramanian,V.Int.J.Hydrog.Energy2014,39(6),2549.doi:10.1016/j.ijhydene.2013.11.075
(16)Lan,J.;Cao,D.;Wang,W.;Ben,T.;Zhu,G.J.Phys.Chem.Lett.2010,1(6),978.doi:10.1021/jz900475b
(17)Ren,H.;Ben,T.;Sun,F.;Guo,M.;Jing,X.;Ma,H.;Cai,K.;Qiu,S.;Zhu,G.J.Mater.Chem.2011,21(28),10348.doi:10.1039/c1jm11307k
(18) Wang,Z.Y.;Li,G.;Sun,Z.G.Acta Phys.-Chim.Sin.2013,29(11),2422.[王朝阳,李 钢,孙志国.物理化学学报,2013,29(11),2422.]doi:10.3866/PKU.WHXB201309021
(19)Xiang,Z.;Cao,D.;Wang,W.;Yang,W.;Han,B.;Lu,J.J.Phys.Chem.C2012,116(9),5974.doi:10.1021/jp300137e
(20)Lan,J.;Cao,D.;Wang,W.;Smit,B.ACS Nano2010,4(7),4225.doi:10.1021/nn100962r
(21)Sun,Y.;Ben,T.;Wang,L.;Qiu,S.;Sun,H.J.Phys.Chem.Lett.2010,1(19),2753.doi:10.1021/jz100894u
(22) Babarao,R.;Dai,S.;Jiang,D.E.Langmuir2011,27(7),3451.doi:10.1021/la104827p
(23)Ahmed,A.;Thornton,A.W.;Konstas,K.;Kannam,S.K.;Babarao,R.;Todd,B.D.;Hill,A.J.;Hill,M.R.Langmuir2013,29(50),15689.doi:10.1021/la403864u
(24) Srinivasu,K.;Ghosh,S.K.J.Phys.Chem.C2011,115(34),16984.doi:10.1021/jp2035218
(25)Wang,W.;Yan,Z.J.;Yuan,Y.;Sun,F.X.;Zhao,M.;Ren,H.;Zhu,G.S.Acta Chim.Sin.2014,72(5),557.[王 维,闫卓君,元 野,孙福兴,赵 明,任 浩,朱广山.化学学报,2014,72(5),557.]
(26)Yuan,Y.;Yan,Z.X.;Ren,H.;Liu,Q.Y.;Zhu,G.S.;Sun,F.X.Acta Chim.Sin.2012,70(13),1446.[元 野,闫卓君,任浩,刘青英,朱广山,孙福兴.化学学报,2012,70(13),1446.]doi:10.6023/A12040104
(27) Manz,T.A.;Sholl,D.S.J.Chem.Theory Comput.2010,6(8),2455.doi:10.1021/ct100125x
(28)Ben,T.;Ren,H.;Ma,S.;Cao,D.;Lan,J.;Jing,X.;Wang,W.;Xu,J.;Deng,F.;Simmons,J.M.;Qiu,S.;Zhu,G.Angew.Chem.Int.Edit.2009,48(50),9457.doi:10.1002/anie.200904637
(29) Frost,H.;Snurr,R.Q.J.Phys.Chem.C2007,111(50),18794.doi:10.1021/jp076657p
(30)Wu,X.J.;Yang,X.;Song,J.;Cai,W.Q.Acta Chim.Sin.2012,70(24),2518.[吴选军,杨 旭,宋 杰,蔡卫权.化学学报,2012,70(24),2518.]doi:10.6023/A12110858
(31) Wu,X.J.;Zheng,J.;Li,J.;Cai,W.Q.Acta Phys.-Chim.Sin.2013,29(10),2207.[吴选军,郑 佶,李 江,蔡卫权.物理化学学报,2013,29(10),2207.]doi:10.3866/PKU.WHXB201307191
(32)Wilmer,C.E.;Farha,O.K.;Bae,Y.S.;Hupp,J.T.;Snurr,R.Q.Energy&Environmental Science2012,5(12),9849.doi:10.1039/c2ee23201d
(33) Wilmer,C.E.;Snurr,R.Q.Chem.Eng.J.2011,171(3),775.doi:10.1016/j.cej.2010.10.035
(34)Wu,D.;Wang,C.;Liu,B.;Liu,D.;Yang,Q.;Zhong,C.AIChE J.2012,58(7),2078.doi:10.1002/aic.v58.7
(35) Schmidt,M.W.;Baldridge,K.K.;Boatz,J.A.;Elbert,S.T.;Gordon,M.S.;Jensen,J.H.;Koseki,S.;Matsunaga,N.;Nguyen,K.A.;Su,S.J.;Windus,T.L.;Dupuis,M.;Montgomery,J.A.J.Comput.Chem.1993,14,1347.
(36) Boys,S.F.;Bernardi,F.Mol.Phys.1970,19,553.doi:10.1080/00268977000101561
(37) Kresse,G.;Furthmuller,J.Comput.Mat.Sci.1996,6,15.doi:10.1016/0927-0256(96)00008-0
(38) Chempath,S.;Clark,L.A.;Snurr,R.Q.J.Chem.Phys.2003,118(16),7635.doi:10.1063/1.1562607
(39) Buch,V.J.Chem.Phys.1994,100,7610.doi:10.1063/1.466854
(40)Wu,X.;Huang,J.;Cai,W.;Jaroniec,M.RSC Adv.2014,4(32),16503.doi:10.1039/c4ra00664j
(41) Peng,D.Y.;Robinson,D.B.Ind.Eng.Chem.Fund.1976,15,59.doi:10.1021/i160057a011
(42) Duren,T.;Sarkisov,L.;Yaghi,O.M.;Snurr,R.Q.Langmuir2004,20(7),2683.doi:10.1021/la0355500
(43)Li,H.;Eddaoudi,M.;Keeffe,M.O.;Yaghi,O.M.Nature1999,402,276.doi:10.1038/46248
(44)Koh,K.;Wong-Foy,A.G.;Matzger,A.J.J.Am.Chem.Soc.2009,131(12),4184.doi:10.1021/ja809985t
(45) Férey,G.;Mellot-Draznieks,C.;Serre,C.;Millange,F.;Dutour,J.;Surblé,S.;Margiolaki,I.Scince2005,309(5743),2040.doi:10.1126/science.1116275
(46)Wong-Foy,A.G.;Matzger,A.J.;Yaghi,O.M.J.Am.Chem.Soc.2006,128(11),3494.doi:10.1021/ja058213h
(47)Yuan,D.;Lu,W.;Zhao,D.;Zhou,H.C.Adv.Mater.2011,23(32),3723.doi:10.1002/adma.v23.32
(48) Farha,O.K.;Yazaydin,A.O.;Eryazici,I.;Malliakas,C.D.;Hauser,B.G.;Kanatzidis,M.G.;Nguyen,S.T.;Snurr,R.Q.;Hupp,J.T.Nat.Chem.2010,2(11),944.doi:10.1038/nchem.834
(49)Yuan,D.;Zhao,D.;Sun,D.;Zhou,H.C.Angew.Chem.Int.Edit.2010,49,5357.doi:10.1002/anie.v49:31
(50) Furukawa,H.;Ko,N.;Go,Y.B.;Aratani,N.;Choi,S.B.;Choi,E.;Yazaydin,A.O.;Snurr,R.Q.;O'Keeffe,M.;Kim,J.;Yaghi,O.M.Science2010,329(5990),424.doi:10.1126/science.1192160
(51) Kokalj,A.Comp.Mater.Sci.2003,28,155.doi:10.1016/S0927-0256(03)00104-6
猜你喜欢
航空材料学报(2023年6期)2023-12-18 05:23:50
小学生学习指导(小军迷联盟)(2023年3期)2023-03-27 09:22:44
中国音乐学(2022年1期)2022-05-05 06:48:46
作物学报(2022年6期)2022-04-08 01:26:24
国际医学放射学杂志(2021年5期)2021-10-22 07:26:20
物理学报(2018年10期)2018-06-14 06:31:32
安徽农业科学(2018年1期)2018-05-14 08:59:41
麦类作物学报(2018年4期)2018-05-11 09:34:08
第一财经(2017年36期)2017-09-25 06:17:11
天然气化工—C1化学与化工(2015年1期)2015-06-01 09:58:37
