
溶菌酶蛋白在聚合物防污膜表面的吸附
2014-02-18 12:07:12胡立梅蔺存国苑世领
物理化学学报 2014年11期
胡立梅 蔺存国 王 利 苑世领
(1山东大学胶体与界面化学教育部重点实验室,济南250100;2海洋腐蚀与防护重点实验室,中国船舶重工集团公司第七二五研究所,山东青岛266101)
1 引言
海洋生物污损会给船舶和海洋设施带来极大的危害:①增加船体自重和船体摩擦阻力,从而增加燃油消耗,增加温室气体排放;②影响船舶的使用性能,如阻塞管道、造成材料结构腐蚀损坏;③增加维护修理费用,带来巨大经济损失.因此,采取有效措施防止污损生物的附着极为重要.其中,涂装防污涂料1-3是最为经济有效的方法.目前广泛使用的防污涂料为以氧化亚铜、硫氰酸亚铜为防污剂的无锡自抛光防污涂料,由于铜元素在港口的累积作用给海洋环境造成严重影响,许多国家已禁止或限制铜类防污剂的使用.因此开展新型对环境友好的防污材料研究势在必行.
依据固体表面与蛋白质之间的相互作用,实验发现亲水性防污涂层材料对抗蛋白质吸附的能力要优于疏水性防污材料,4,5归因于蛋白质内部多为疏水性残基,因此疏水性防污膜对蛋白质具有更大的亲和性;同时,亲水性材料表面稳定的水化层对蛋白质的吸附过程也具有能障作用,6-9因此亲水涂层对蛋白质吸附也具有防污性能.亲水或疏水材料表面不同的结构和化学性质,会影响周围水分子的聚集行为和蛋白质的吸附,引起抗污损性能的差异,其防污机理也有所不同.
聚乙二醇(PEG)是一种非枝化水溶性高分子材料,与许多物质有很好的相容性,具有良好的稳定性和润滑性,是目前应用最为广泛、抗污染效果最好的防污材料之一.10,11因而,PEG常被作为“标尺”,衡量其他材料的抗污染性能.在新兴众多环保型海洋防污新材料中,以聚二甲基硅氧烷(PDMS)为代表的有机硅聚合物涂层由于具有较低的表面能、不粘性和在水中的稳定性,在海洋船舶中的应用越来越广泛.12,13在众多实验对比研究中,多把PEG聚合物膜归为亲水防污涂层,而把PDMS聚合物膜当作疏水涂层,通过对比两种防污材料的结构差异和防污机理,将有助于理解不同结构类型防污涂层对蛋白质吸附的影响.
实验研究推测,材料的防污能力与其表面结合的水化层密切相关,但是如何从微观层次上理解防污材料表面水化层所起的作用,传统实验研究还有一定的难度,其中蛋白质吸附的微观过程一直是实验研究的难点,也是蛋白质构象转换研究的制约因素.利用分子动力学模拟14在分子层次上研究蛋白质与材料表面的相互作用,阐释相关吸附机理,是对宏观实验的一项有益补充,对理解防污材料表面的水化层结构,以及材料表面微观结构与宏观防污能力之间的关系有不可替代的作用.本文通过分子动力学方法研究了溶菌酶蛋白在聚合物防污材料(PEG和PDMS)表面的吸附行为,通过分析蛋白质与两种聚合物结合位点、蛋白质与吸附膜的作用能,以及表面膜附近水分子的氢键和动力学性质等,寻找亲水和疏水两种聚合物膜防污性能差异的原因,从微观层面上阐述防污材料的防污机理.
2 模型与方法
2.1 模型
模拟中选择溶菌酶蛋白作为目标分子,研究蛋白质在防污涂层上的吸附.含有129个氨基酸的溶菌酶蛋白具有优良的二级结构,存在典型的α螺旋和β折叠结构,易于在模拟过程中观察其结构变化.大量的实验和理论方法都对溶菌酶分子的结构、空间折叠等进行过讨论,15,16对其详细结构的认识有利于我们讨论防污膜与蛋白质之间的相互作用.模拟中溶菌酶蛋白的晶体结构从蛋白质数据库中获得(PDB号为7LYZ).选取的两种聚合物膜包括:(1)利用Materials Studio程序包中的Amorphous Cell模块构建密度为0.965 g∙cm-3,厚度为2 nm的PDMS基底;(2)从Materials Studio原有数据库中调出PEG晶格,17分别在x,y,z方向上扩展得到与PDMS基底面积相近的PEG基底,并确保蛋白质分子在其表面上有足够大的接触面积.溶菌酶分子带有8个正电荷,因此在模拟体系中加入8个氯离子作为抗衡离子以平衡体系电荷.为充分研究聚合物膜与蛋白质在水中的相互作用,我们在聚合物膜上方分别添加了相同厚度的水层.作为初始构型将溶菌酶分子以相同方向和位置放在距聚合物膜0.7 nm17处的水相中.同时为防止构象重叠,删除蛋白质和聚合物膜周围0.2 nm之内的水分子.
2.2 模拟方法
模拟采用GROMOS 45a3联合原子力场,18选用GROMACS 4.0.5进行分子动力学计算.19,20模拟中采用周期性边界条件,其中PDMS基底体系的格子大小选为12.8 nm×10 nm×20 nm,PEG基底体系为13 nm×10 nm×20 nm.在模拟过程中,PEG和PDMS基底的底部固定,表面膜部分未加约束条件,使模拟结果更加准确.聚合物膜所用的力场参数及电荷参见表1.模拟中溶剂水模型为单点电荷(SPC)模型,21采用particle-mesh Ewald(PME)方法22处理长程静电相互作用,非键相互作用的截断半径选择1.2 nm.初始构型建立以后,对所有体系首先利用最速下降法进行能量优化以消除可能的构象重叠,随后进行分子动力学方法计算.模拟中选择Berendsen热浴法23控制温度,采用LINCS方法24约束键长.分子动力学模拟阶段,固定蛋白质质心在298 K温度下进行15 ns的分子动力学模拟;然后取消对质心的固定,再进行20 ns的动力学模拟,让蛋白质在自由状态下与聚合物膜作用达到平衡,其中模拟步长为2 fs,间隔0.2 ps记录一次轨迹信息.
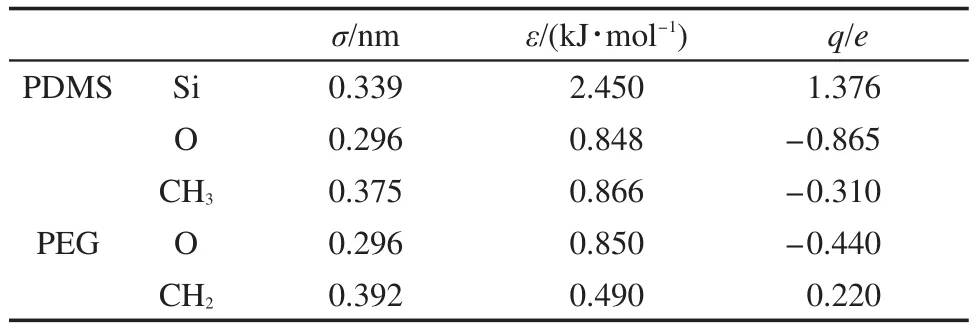
表1 模拟中使用的PDMS和PEG的力场参数Table 1 Force field parameters for PDMS and PEG used in this work
3 结果与讨论
3.1 溶菌酶蛋白在聚合物膜表面吸附行为
通过整个模拟过程发现随着模拟时间的变化,溶菌酶蛋白均会吸附到两种聚合物表面,在吸附过程中溶菌酶蛋白构型及性质有不同程度的变化.从图1中可以看到蛋白质在两种聚合物膜表面吸附后的构型形态不同且吸附在膜表面的残基也不同.在PDMS表面,蛋白质倾向于平铺吸附,吸附在表面的残基数相对较多;而在PEG表面,蛋白质是以侧立的方式吸附于PEG表面,与膜的接触面积较小.这是因为蛋白质中极性氨基酸与非极性氨基酸在疏水表面和亲水表面相互作用方式不同造成的.疏水基团的空间位阻使得分子取向受到限制,尽管在水溶液中蛋白质具有将疏水基团折叠在分子内部,表面显露极性和带电基团相互作用,但总有一些疏水性基团或极性基团的疏水部位暴露在蛋白质表面.这部分疏水基团可与弱疏水基团发生疏水相互作用,被疏水性吸附剂所吸附,因此疏水性表面易于蛋白质分子的吸附.
为了更好地了解两种表面对蛋白质的吸附特性,统计了吸附在聚合物膜表面上氨基酸残基(表2).总体上,在PDMS表面上吸附的氨基酸数目要比在PEG表面上的多,同时,在PDMS表面吸附的氨基酸类型中非极性氨基酸所占的比例要比在PEG表面上的高,亲水指数要大.通常,亲水指数越大,疏水性就越强,亲水性就越弱.我们推测这是由于PDMS相对PEG为疏水结构.从表2中可以看出溶菌酶蛋白回转半径(Rg)相对于在纯水中有变化:在PDMS膜表面有所增大,而在PEG膜表面有所减小.一般而言,蛋白质内部多为疏水残基而表面多为亲水残基,因此蛋白质倾向于将内部的疏水残基翻转从而与PDMS更好地通过疏水相互作用结合,吸附在PDMS表面蛋白质中心结构较稀疏,这是蛋白质内部的残基翻转的结果,同时吸附在膜表面的面积较大.而PEG表面吸附的蛋白质残基的平均亲水指数较小,归因于PEG是亲水表面,因而蛋白质更倾向于通过表面的亲水残基与PEG作用.从图2中可以看出,吸附在PEG膜表面的蛋白质相对紧凑,几乎没有翻转,同时与膜接触的面积较小.
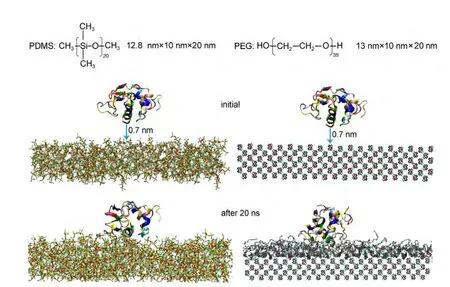
图1 溶菌酶蛋白在聚合物(PDMS,PEG)膜表面的初始构型及20 ns动力学模拟之后溶菌酶吸附在聚合物膜表面的形态Fig.1 Initial configurations of lysozyme protein above the non-fouling membranes and snapshots oflysozyme′s adsorption on two polymer(PDMS,PEG)membranes after 20 ns dynamics simulation

表2 20 ns动力学模拟过程中吸附的残基类型及蛋白质参数的变化Table 2 Adsorbed residue types and changes of protein parameters during 20 ns dynamics simulation
蛋白质分子的均方根偏差(RMSD)可以用来表征其构象变化,RMSD是关于分子在模拟过程中运动幅度的一个衡量,RMSD值越大,分子在模拟过程中的位置变化越剧烈.RMSD的定义为:
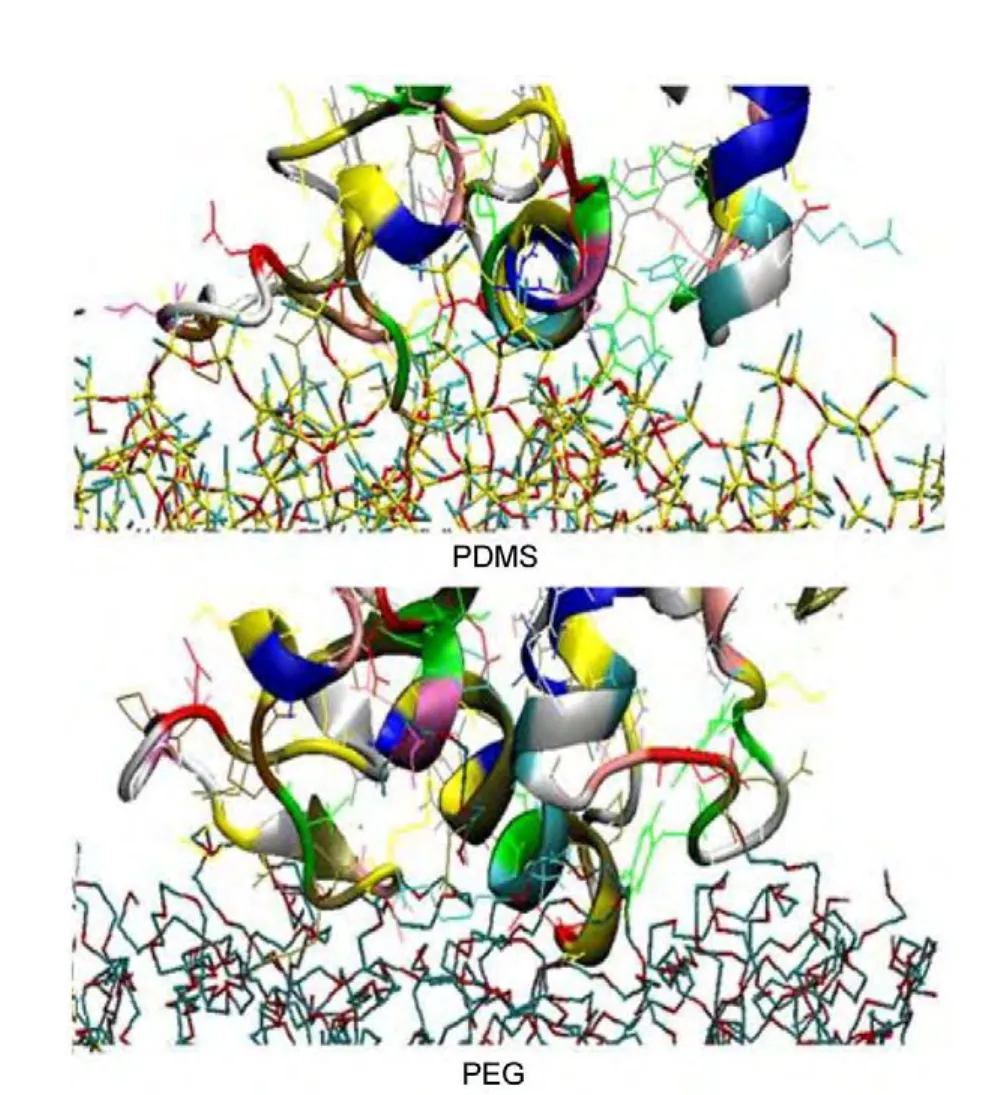
图2 20 ns动力学模拟之后溶菌酶吸附在两个聚合物膜表面的形态及吸附残基Fig.2 Snapshots and adsorbed residues oflysozyme′s adsorption on two polymer membranes after 20 ns dynamics simulation
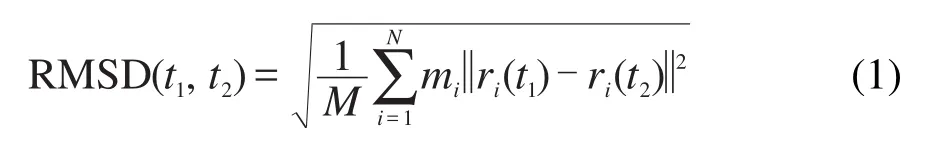
其中mi是原子i的质量,M为所有mi的和,ri(t)是原子i在时间t时的位置.t1为模拟过程中的某一时刻,t2一般为初始时刻,t2=0.在本工作中,溶菌酶分子的初始结构作为RMSD计算的参考结构.我们计算了溶菌酶分子中所有原子的RMSD值以及骨架碳的RMSD值(表2).在PDMS表面与PEG表面上溶菌酶分子的Cα-RMSD值均比溶液中的值有所增加,表明表面诱导了蛋白质分子构象发生变化.其中,在PDMS表面上Cα-RMSD值要比在PEG表面上的值大,说明溶菌酶蛋白在PDMS表面上的构型变化比较大.两个表面上所有原子的RMSD值要比Cα-RMSD值变化大,这表明溶菌酶分子的侧链、环区域或序列末端有较大的运动.同时,从RMSD随模拟时间的变化(图3)显示RMSD值的增加主要在3.0 ns前,表明构象的变化主要是在蛋白质以优势取向吸附在表面后发生的,这与蛋白质吸附的“两个阶段”机理一致.第一个阶段(快速吸附阶段)主要是短时间尺度的取向变化阶段,蛋白质通过平移和转动运动优化其与表面的相互作用,并以优势取向吸附在表面.第二个阶段(缓慢吸附平衡阶段)为长时间尺度的表面诱导蛋白质吸附构象变化阶段.
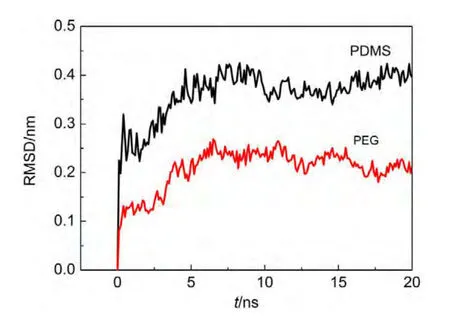
图3 溶菌酶均方根偏差(RMSD)随时间的变化Fig.3 Root mean square deviation(RMSD)of lysozyme changes over time
3.2 吸附过程中能量的变化
蛋白质与表面的相互作用力主要是范徳华力和库仑引力等非键作用力.对于非键作用能包含三项:排斥项、吸引项、库仑项.范德华作用的排斥项和吸引项的作用能用Lennard-Jones势能表示,而库仑项则用库仑作用能表示.溶菌酶蛋白质分子中具有疏水的、非极性的部分,可以通过范德华力直接与材料表面相互吸引.而蛋白质分子中带有8个正电荷,电荷的存在使其与膜表面之间形成库仑相互作用.从图4中了解到,不管是范德华作用能还是库仑作用能,PDMS体系中的能量要比PEG体系中的能量高,基底对蛋白质的强的作用能表明PDMS基底对溶菌酶蛋白有较强的吸附作用.
我们分析了两个体系在整个过程中的总非键相互作用能的变化,从图4中可以看到,在整个蛋白质与表面的相互作用过程中,溶菌酶蛋白与PDMS膜表面的结合能要比PEG膜表面的结合能要高,这表现为PDMS膜表面对溶菌酶蛋白分子具有更强的相互作用,强的相互作用牵引蛋白质吸附,吸附后构型也更加稳定,不易脱离.同时,研究了范德华相互作用与库仑相互作用在整个作用能中所占比例,发现在两个体系中,范德华作用均贡献了大部分.文献资料25认为,由于是由取向性不好的范德华作用占主导,蛋白质吸附时的取向性也是随机的.
3.3 水化层分子
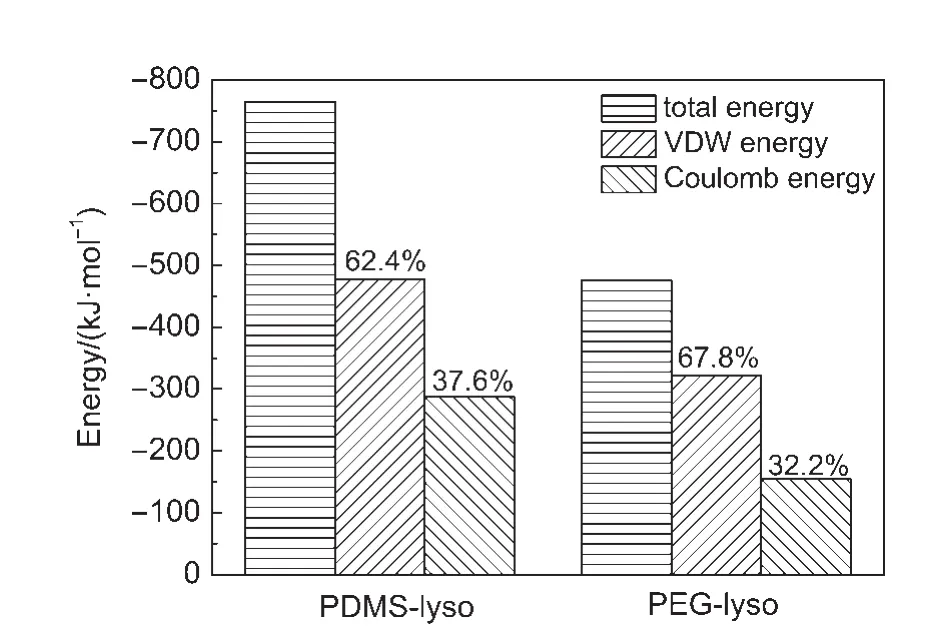
图4 溶菌酶与聚合物膜中范德华作用能和库仑作用能所占的比例Fig.4 Decomposed van der waals(VDW)and Coulomb energies between lysozyme and polymer membranes
目前关于蛋白质与膜表面的相互作用中,普遍的观点26,27均认为其与聚合物表面紧密结合的水化层有关,蛋白质要吸附在聚合物膜表面首先要克服该水化层形成的物理壁垒和能量障碍.我们将距离界面膜原子在0.5 nm范围内的水分子定义为水化层水分子,通过水化层中水分子的动力学行为可以了解在蛋白质吸附到聚合物表面过程中水化层分子所起作用.
3.3.1 水化层与聚合物膜间的均力势
为对比两种聚合物膜对水分子的束缚能力,我们采用均力势(PMF)来表示聚合物膜表面的水化层的能障作用.模拟中选取界面膜表层原子与水分子之间的径向分布函数g(r),通过方程E(r)=-kBTlng(r)求得PMF,其中kB是波尔兹曼常数,T是温度,其变化曲线如图5所示,表示外部水分子靠近聚合物膜的过程中所要克服的阻碍作用.以PEG体系为例说明如下几点:(1)势能曲线极小值点(CM)大约在0.20 nm处,表示聚合物膜和一个水分子直接接触的距离;(2)势能曲线上的第二个极小值点大约在0.41 nm处,是溶剂分离极小点(SSM),为第二个溶剂层与水分子接触的位置,CM和SSM对应的能量数值决定了第一水化层和第二水化层中水分子与聚合物膜结合的稳定性;(3)CM和SSM中间是一个比较高的能垒,也就是溶剂层能障(BS),表示水分子从聚合物膜的第二水化层进入第一水化层,与聚合物膜结合时所需要克服的水化层分子的阻碍作用.随着水分子与聚合物膜间距离的增大,在无穷远处PMF趋向于0.
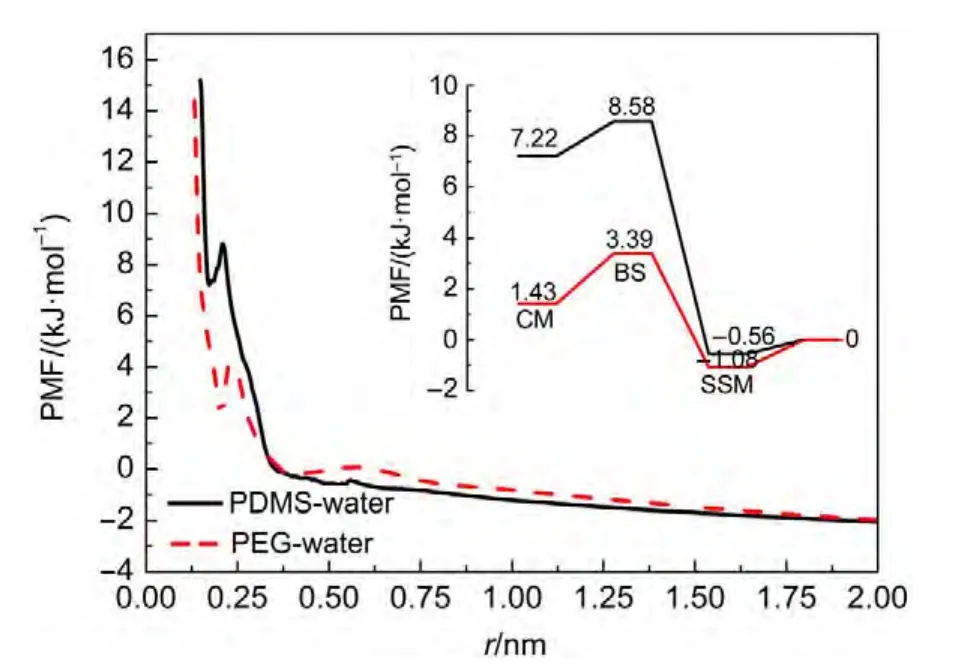
图5 水分子与聚合物膜间的均力势(PMFs)Fig.5 Potential of mean forces(PMFs)between water and polymer membranes
聚合物膜和水化层分子之间的结合能ΔE+=从图5中可以看出:(1)体系II(PEG基底-蛋白质)中PEG聚合物膜与水分子之间形成了能量上更加稳定的水化层,对应第一水化层中能量数值为1.43 kJ∙mol-1,远小于体系I(PDMS基底-蛋白质)中PDMS聚合物膜与水分子之间的能量数值(7.22 kJ∙mol-1);(2)体系II中PEG聚合物膜与水分子之间的结合能ΔE+(4.47 kJ∙mol-1)要小于体系I中PDMS聚合物膜与水分子之间的结合能ΔE+(9.14 kJ∙mol-1),表示水分子PEG聚合物膜之间的结合需要克服的能垒要低,即水分子与PEG之间的结合相对而言要容易一些;但是其解离能ΔE-(1.96 kJ∙mol-1)要大于体系I中PDMS聚合物膜与水分子之间的解离能ΔE-(1.36 kJ∙mol-1),亦即相对体系I而言,水分子易于与PEG结合,但是当形成稳定的结合对以后,水分子亦不易解离.上述分析表明,相对PDMS膜而言,水分子易于跨过水化层分子的阻碍作用与PEG聚合物膜相结合,但是二者结合后也不易解离,说明PEG聚合物膜与水分子的结合更牢固.
3.3.2 水化层分子的扩散性质
为了更好地研究水化层分子的作用,我们将溶菌酶固定质心且距膜表面1.0 nm进行了20 ns的动力学模拟.计算了模拟过程中最后5 ns聚合物膜表面水化层分子的均方位移(MSD)-时间曲线,如图6所示.从图6中可以看出均方位移与时间呈线性关系,说明模拟已经达到平衡.通过公式(2)可求得水化层分子的扩散系数(D):

图6 两聚合物膜表面水化层分子的均方位移(MSD)-时间曲线Fig.6 Plots of mean square displacement(MSD)vs time of hydration molecules above two polymer membranes
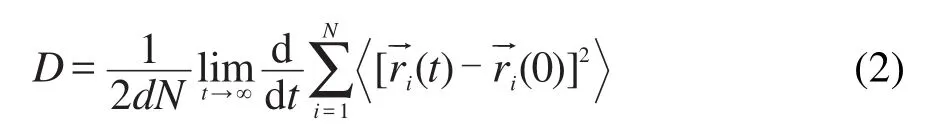
其中d为维度数,等于3,N为水化层中的水分子数,分别为第i个粒子在时刻t和0时的坐标,即为图6中直线的斜率.我们分析了聚合物膜表面与水花层分子之间形成的氢键数目及氢键寿命(表3),通过计算发现单位面积上PEG与水分子形成的氢键数约为1.2 nm-2,而PDMS单位表面积上与水分子形成的氢键数约为0.36 nm-2.通过与体相水的比较,发现水分子在两种膜表面的扩散均呈一定程度的降低,这是由于膜与水之间存在较强的相互作用,并且PEG由于和水化层分子间存在更多的氢键和静电相互作用,因而扩散更慢,说明聚合物膜将该水化层分子紧密束缚在表面.
3.3.3 弛豫时间
界面附近水分子的弛豫时间能很好地反映界面与水化层的关系,弛豫时间越长说明界面对水层的束缚能力越强.弛豫时间通常由在一个时间步长内残留在初始厚度范围内水的比例来决定,弛豫时间可通过寿命自相关函数Cr(t)计算得到:28-30
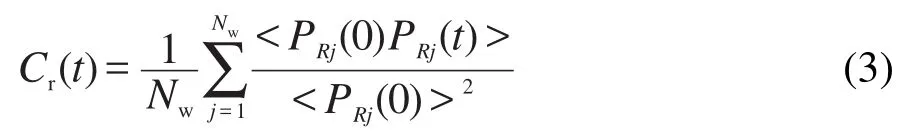
其中PRj是一个二值算符(R指水化层分子的厚度),如果第j个水分子在t时刻时仍停留在初始选定的区域内,则PRj(t)=1,否则PRj(t)=0.Nw是所有在初始选定范围内的水分子数,<>代表系综平均.
模拟中选取距离聚合物膜0.4 nm水化层内的水分子进行分析,其Cr(t)-t关系图如图7所示.为了定量地表示弛豫时间,对寿命自相关函数进行指数拟合,Cr(t)=Arexp(-t/τr),其中Ar表示强度系数,τr为弛豫时间.拟合得到的两种聚合物膜附近水的弛豫时间见表3.同样体相水的弛豫时间一并列出.从表3中可以发现,PDMS和PEG表面水的弛豫时间都比体相水长,说明PDMS和PEG均对水分子有束缚作用,限制了水分子逃逸出水化层的运动,而PEG表面的这种限制作用更加明显.这表明PEG表面结合的水化层分子更加紧密,而蛋白质要穿过这层水化层分子吸附到表面的难度就更大.

表3 两表面水化层分子的扩散系数(D),弛豫时间(τr)和氢键性质Table 3 Diffusion coefficients(D),residence times(τr),and H-bond of hydration molecules of the two surfaces
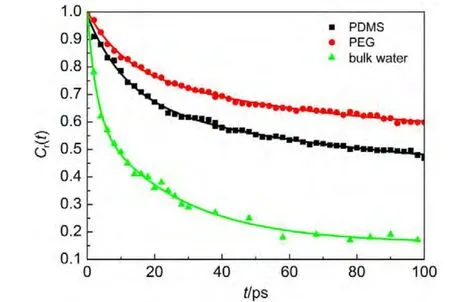
图7 水化层分子的寿命自相关函数(Cr(t))Fig.7 Survival time correlation functions(Cr(t))of hydration molecules
3.4 疏水/亲水膜的防污机理
蛋白质在聚合物膜表面的吸附是一个膜与蛋白作用,和膜与水化层分子作用之间相互竞争的结果.首先蛋白质和聚合物膜在溶剂环境中会在表面极化出一层紧密结合的水化层,蛋白质要在膜表面吸附首先就要使膜表面和蛋白质表面去溶剂化.该过程需要克服一定的能量势垒,然后才能在膜与蛋白质间作用力的驱动下,蛋白质逐渐接近膜表面.从文中的分析可以看出,蛋白质与亲水性膜和疏水性膜的相互作用方式是不同的.在疏水性膜表面,其对水化层分子的束缚作用较弱,蛋白质吸附到膜表面需要克服的能量较低.同时,由于蛋白质分子内部多为疏水性残基,在靠近疏水性膜的过程中,蛋白质分子会在膜表面原子的诱导下发生构象翻转,使内部的疏水残基暴露(图8a),与膜表面通过疏水相互作用更紧密的结合,接触面积较大(图8b),吸附相对更加稳定;在亲水性膜表面,由于膜与水化层分子的结合较为紧密(图8c),蛋白质要吸附到膜表面需要克服更高的能量,蛋白质分子表面多为亲水残基,所以在于亲水性膜表面的相互作用过程中,蛋白质构象翻转不大,吸附后与表面膜的接触面积相对较小(图8d).

图8 蛋白质在疏水性与亲水性聚合物膜的吸附行为示意图Fig.8 Illustration of protein adsorption on hydrophobic and hydrophilic polymer membranes
4 结论
本文对溶菌酶蛋白在聚二甲基硅氧烷和聚乙二醇表面的动力学行为进行了研究.经过20 ns的动力学模拟,蛋白质在两种防污膜表面均完成了吸附,并且在更长的时间范围内也未完成脱附.通过对蛋白质在表面的构像变化、能量变化以及两个表面的水化层分子的研究表明:(1)对比两体系中的RMSD及Rg的变化,发现蛋白质在PDMS表面翻转变形较大,且在PDMS表面上吸附的残基类型及数目较多.这主要是因为相对PEG表面,PDMS为疏水性聚合物膜,蛋白质在疏水性材料上容易将内部的疏水残基翻转出来更好地与疏水表面通过疏水相互作用结合.(2)PDMS表面对蛋白质的吸附能较高,其中范德华作用能占主导作用.较高的作用能牵引蛋白质吸附到材料表面,不宜于蛋白质防污应用.(3)我们从扩散性质和弛豫时间对水化层分子进行分析,结果表明水化层分子在PDMS表面的扩散系数较大,弛豫时间较短,即PDMS表面对水化层分子的束缚作用较弱.这是由于首先PEG相对于PDMS表面有着更丰富的氢键受体,单位面积上形成的氢键数目及氢键的寿命较长;其次,更为重要的是PEG表面形成的氢键寿命更长,即其与水分子形成的氢键不易断裂,使得PEG与水分子的结合更加紧密,降低了表面水分子的可运动性,形成了一层结合稳定的水化层,能够很好地阻碍蛋白质的吸附.综上所述,本文通过研究蛋白质与PEG和PDMS聚合物膜的相互作用,从微观角度解释了PEG表面相对PDMS表面阻抗蛋白质吸附性能更佳的原因.
(1)Ostuni,E.;Chapman,R.G.;Holmlin,R.K.;Takayama,S.;Whitesides,G.M.Langmuir2001,17,5605.doi:10.1021/la010384m
(2)Shen,M.C.;Martinson,L.;Wagner,M.S.;Castner,D.G.;Ratner,B.D.;Horbett,T.A.J.Biomater.Sci.Polym.Ed.2002,13,367.doi:10.1163/156856202320253910
(3) Chambers,L.D.;Stokes,K.R.;Walsh,F.C.;Wood,R.J.Surface and Coatings Technology2006,201(6),364.
(4) Zheng,J.;Li,L.;Chen,S.;Jiang,S.Langmuir2004,20(20),8931.doi:10.1021/la036345n
(5) Cedervall,T.;Lynch,I.;Foy,M.;Berggård,T.;Donnelly,S.C.;Cagney,G.;Dawson,K.A.Angewandte Chemie International Edition2007,46(30),5754.
(6)Aggarwal,P.;Hall,J.B.;McLeland,C.B.;Dobrovolskaia,M.A.;McNeil,S.E.Advanced Drug Delivery Reviews2009,61(6),428.doi:10.1016/j.addr.2009.03.009
(7) Kitano,H.;Sudo,K.;Ichikawa,K.;Ide,M.;Ishihara,K.The Journal of Physical Chemistry B2000,104(47),11425.doi:10.1021/jp000429c
(8)Zheng,H.R.;Wang,X.W.;Lin,X.H.;Geng,Q.;Chen,X.;Dai,W.X.;Wang,X.X.Acta Phys.-Chim.Sin.2012,28,1764.[郑华荣,王晓韡,林霞晖,耿 强,陈 旬,戴文新,王绪绪.物理化学学报,2012,28,1764.]doi:10.3866/PKU.WHXB201205112
(9) Lüsse,S.;Arnold,K.Macromolecules1996,29(12),4251.doi:10.1021/ma9508616
(10)Wang,Y.Q.;Wang,T.;Su,Y.L.;Peng,F.B.;Wu,H.;Jiang,Z.Y.Langmuir2005,21(25),11856.doi:10.1021/la052052d
(11) Zhao,W.;Su,Y.;Li,C.;Shi,Q.;Ning,X.;Jiang,Z.Journal of Membrane Science2008,318(1),405.
(12) Chen,H.;Yuan,L.;Song,W.;Wu,Z.;Li,D.Progress in Polymer Science2008,33(11),1059.doi:10.1016/j.progpolymsci.2008.07.006
(13) Michalkova,A.;Tulyani,S.;Beals,J.;Leszczynski,J.Journal of Molecular Modeling2012,18(1),239.doi:10.1007/s00894-011-1058-8
(14)Shen,J.W.;Wu,T.;Wang,Q.;Pan,H.H.Biomaterials2008,29(5),513.doi:10.1016/j.biomaterials.2007.10.016
(15) Bai,S.;Li,H.;Zhang,L.Acta Phys.-Chim.Sin.2013,29,849.[白 姝,李 浩,张 麟.物理化学学报,2013,29,849.]doi:10.3866/PKU.WHXB201301182
(16)Fang,Y.Y.;Hu,X.G.;Yu,L.;Li,W.B.;Lin,R.S.Acta Phys.-Chim.Sin.2007,23,84.[方盈盈,胡新根,于 丽,李文兵,林瑞森.物理化学学报,2007,23,84.]doi:10.3866/PKU.WHXB20070117
(17)Panos,M.;Sen,T.Z.;Ahunbay,M.G.Langmuir2012,28(34),12619.doi:10.1021/la301546v
(18) Schuler,L.D.;Daura,X.;Van Gunsteren,W.F.Journal of Computational Chemistry2001,22(11),1205.
(19) Van der Spoel,D.;Lindahl,E.;Hess,B.;Groenhof,G.;Mark,A.E.;Berendsen,H.J.Journal of Computational Chemistry2005,26(16),1701.
(20) Hess,B.;Kutzner,C.;Van der Spoel,D.;Lindahl,E.Journal of Chemical Theory and Computation2008,4(3),435.doi:10.1021/ct700301q
(21) Lindahl,E.;Hess,B.;Van der Spoel,D.Molecular Modeling Annual2001,7(8),306.
(22) Essmann,U.;Perera,L.;Berkowitz,M.L.;Darden,T.;Lee,H.;Pedersen,L.G.The Journal of Chemical Physics1995,103(19),8577.doi:10.1063/1.470117
(23) Berendsen,H.J.;Postma,J.P.M.;van Gunsteren,W.F.;DiNola,A.R.H.J.;Haak,J.R.The Journal of Chemical Physics1984,81(8),3684.doi:10.1063/1.448118
(24) Hess,B.;Bekker,H.;Berendsen,H.J.;Fraaije,J.G.Journal of Computational Chemistry1997,18(12),1463.
(25) Dismer,F.;Hubbuch,J.Journal of Chromatography A2007,1149(2),312.doi:10.1016/j.chroma.2007.03.074
(26) Hower,J.C.;He,Y.;Bernards,M.T.;Jiang,S.The Journal of Chemical Physics2006,125(21),214704.doi:10.1063/1.2397681
(27) Vanderah,D.J.;La,H.;Naff,J.;Silin,V.;Rubinson,K.A.Journal of the American Chemical Society2004,126(42),13639.doi:10.1021/ja047744n
(28) Shao,Q.;He,Y.;White,A.D.;Jiang,S.The Journal of Physical Chemistry B2010,114(49),16625.doi:10.1021/jp107272n
(29) He,Y.;Hower,J.;Chen,S.;Bernards,M.T.;Chang,Y.;Jiang,S.Langmuir2008,24(18),10358.doi:10.1021/la8013046
(30) He,Y.;Chang,Y.;Hower,J.C.;Zheng,J.;Chen,S.;Jiang,S.Physical Chemistry Chemical Physics2008,10(36),5539.doi:10.1039/b807129b
猜你喜欢
生物化学与生物物理进展(2022年6期)2022-07-21 11:52:06
成都大学学报(自然科学版)(2021年1期)2021-05-22 01:31:18
中学生数理化(高中版.高考理化)(2021年2期)2021-03-19 08:52:38
理化检验-化学分册(2020年12期)2021-01-26 00:41:40
池州学院学报(2015年3期)2016-01-05 01:13:04
天津科技大学学报(2015年2期)2015-08-09 01:40:42
体育世界(学术版)(2015年3期)2015-07-01 17:15:41
应用化工(2014年11期)2014-08-16 15:59:13
中国海洋大学学报(自然科学版)(2014年6期)2014-02-28 12:21:01
食品科学(2013年14期)2013-03-11 18:25:13
