
Ce-Cu-Co/CNTs催化剂催化合成气制低碳醇及乙醇的研究
2014-02-18 12:07:08王晓东2刘双强游向轩
物理化学学报 2014年11期
韩 涛 黄 伟*, 王晓东2 唐 钰 刘双强 游向轩
(1太原理工大学煤科学与技术教育部和山西省重点实验室,太原030024;2中国科学院山西煤炭化学研究所煤转化国家重点实验室,太原030001)
1 引言
随着燃料需求的不断增长和石油资源的日益枯竭,积极寻求和开发新的能源体系已是迫在眉睫.由合成气(CO+H2)制取低碳混合醇(C1-C6)是煤炭资源洁净利用的重要途径之一.低碳混合醇不仅可以作为替代燃料和清洁汽油添加剂,还可以作为化学品和化工原料,近年来在燃料、化工和环保领域的应用价值逐步凸现,相关研究也日益活跃.1-3乙醇属于低碳醇之一,是大宗化工原料,由乙醇出发可以生产许多高附加值的化工产品,如乙酸乙酯、乙醇胺和乙烯等.同时,相对于甲醇而言,乙醇的热值更高且无毒,使用乙醇作为汽油添加剂生产乙醇汽油,不仅可以增加汽油的内氧,使之燃烧充分,达到节能环保的目的,而且还可以提高汽油的抗爆性(辛烷值).在世界面临石油燃料短缺的今天,开发利用乙醇燃料,受到人们越来越多的广泛关注.4
CuCo基催化剂由于反应条件温和,活性高,碳链增长能力强而被认为是最具有工业应用前景的低碳醇合成催化剂.但由于反应过程中常伴随着烃合成、甲醇合成以及水煤气变换等多个副反应,导致总醇选择性不高,5-11醇产物碳数分布宽.这不仅增加了分离难度,而且也严重影响了整体工艺的经济性.
碳纳米管具有高的比表面积、优良的电子传导性以及良好的热稳定性和吸脱附性能,是一种新型的催化剂载体或助剂.12-14碳纳米管特殊的孔腔结构对于其中的金属粒子具有限域作用,从而能够起到缩窄产物分布、提高某种碳数产品选择性的作用.15
本文将Cu-Co基催化剂高活性和高碳链增长能力与碳纳米管的限域效应结合起来,通过添加不同量的Ce来改善活性金属粒子的晶粒尺寸,增加合成醇活性位的数量,11,16达到增加醇产物选择性和实现对产物碳数分布的控制.相比于文献报道,11,13,17,18本文催化剂缩窄了产物分布,大幅度提高了乙醇的选择性.
2 实验部分
2.1 催化剂的制备
催化剂制备采用共浸渍法,使用的碳纳米管为经过机械剪切后端口打开的短碳纳米管(表示为s-CNTs,0.5-2 μm),这种碳纳米管有利于在浸渍时金属粒子进入碳纳米管内部.具体步骤如下:称取一定比例的Cu(NO3)2∙3H2O、Co(NO3)2∙6H2O(AR,国药集团化学试剂有限公司)和Ce(NO3)3∙6H2O(AR,天津市科密欧化学试剂开发中心)配制成混合水溶液,Cu、Co质量比为2:1,总负载量为18%.然后将适量s-CNTs(中国科学院成都有机化学有限公司)浸渍于上述混合溶液中,经充分搅拌后,放入110°C烘箱中过夜,再在氮气氛围中400°C焙烧4 h,制得的催化剂用Ce-Cu-Co/CNTs表示,Ce的质量分数分别为0%、1%、3%和5%,为表述方便,所制备的催化剂分别表示为Cx,x代表Ce的质量百分数.
2.2 催化剂的表征
催化剂的XRD分析采用日本RigakuD/max-2500型衍射仪,CuKα辐射,Ni滤片,管电压40 kV,电流30 mA,连续扫描法,扫描范围2θ=10°-90°,扫描速率8(°)∙min-1.
催化剂的H2-TPR表征在天津先权应用技术研究所生产的TP-5000吸附仪上进行.H2-TPR表征催化剂用量为50 mg,以5%H2-95%N2为还原气,流速为20 mL∙min-1,以10 °C∙min-1的速率由50 °C升到800°C,热导检测耗氢量,测定TPR曲线.用20 mg Ag2O还原耗氢量作为外标计算定量基准.
催化剂的比表面积与孔径分布采用JWBK122W型物理吸附仪测定,测试前样品在300°C真空脱气3 h,于液氮温度(-197°C)下进行N2吸/脱附实验,用Brunauer-Emett-Teller(BET)方程计算样品比表面积,用Barret-Joyner-Halenda(BJH)方法计算孔径分布.
TEM样品的TEM数据是通过JEOL-2100F型透射电镜在200 kV条件下获得的.
催化剂的CO-TPD表征在Micromeritics Autochem 2920吸附仪上进行.催化剂用量为100 mg,室温下通入10%H2-90%He的混合气,以10°C∙min-1的速率升温至400°C,维持1 h,然后降温至50°C,通入10%CO-90%He的混合气,在50°C吸附至饱和后,切氦气吹扫至基线平稳后,以10°C∙min-1的升温速率进行脱附.
2.3 催化剂的活性评价
催化剂性能的评价是在加压微型固定床反应器(内径为8 mm,高240 mm)中进行;催化剂填装量为1 g;反应前催化剂用含30%H2和70%N2的混合气体对催化剂进行程序升温还原,以2°C∙min-1的升温速率升至400°C,并在该温度下还原2 h后,在N2流中降温至反应温度(300°C)并稳定后,切换成合成气(H2/CO体积比为2),缓慢升压至1.5 MPa后进行连续反应,并开始采样.反应产物利用日本岛津公司生产的GC-14B气相色谱仪(色谱柱:Porapak填充柱)在线分析.
3 结果与讨论
3.1 活性评价结果
CuCo低碳醇合成催化剂最初由法国石油研究所发明,其后被大量地研究.19,20该催化剂通常以Cu、Co、K为基本成分,辅以一种或两种非还原氧化物(Cr2O3,Al2O3,ZnO等),以及一种或若干种助剂(稀土氧化物、贵金属、Sc、Yb、Zr、Th等).近十余年来随着碳纳米管规模化制备的实现和对其电传导性质的认识,碳纳米管亦被广泛用作催化剂助剂和载体,在低碳醇的合成中也得到了应用,并展现出一定的促进作用.12-14Dong等13用共沉淀法制备了“人字形”多壁碳纳米管改性的CuCo基催化剂,在5.0 MPa,573 K,空速(GHSV)=7200 mL∙g-1∙h-1的反应条件下,DME+C2-8醇的时空收率达到760 mg∙g-1∙h-1,C2-C8醇选择性为57.9%,其中丁醇所占比例最大,占20%,乙醇只有不到10%.Wang17和Shi18等分别采用初湿共浸渍法和超声浸渍法制备了CuCo/CNTs催化剂,得到相对较窄的醇分布,但是醇中甲醇最多,可达54%以上,且催化剂活性较低.
稀土金属因其特有性质,越来越多地作为助催化剂使用.士丽敏9和Pan15等在研究La、Ce的添加对超声辅助的反向共沉淀法制备的CuCo基催化剂的影响时发现,La、Ce的添加能够显著地降低材料的晶粒尺寸,提高催化剂比表面积,促进合成醇活性位的形成.毛东森等11发现,当Ce含量为10%时,共浸渍法制备的CuFe/SiO2催化剂的醇的时空收率可达121.0 g∙kg-1∙h-1,比未添加Ce的催化剂的时空收率提高一倍以上.
从上述研究中可以看出,采用共沉淀法制备的碳纳米管担载CuCo催化剂尽管有较高的活性,但由于共沉淀法自身特点使活性组分不能有效进入碳纳米管孔腔内,限域效应未能体现,因此产物分布宽没有得到改善.13而已有的浸渍法碳纳米管担载CuCo催化剂,由于没有考虑多组分浸渍易造成不均匀性以及金属Cu塔曼温度低,在干燥、焙烧和反应过程中易于烧结等因素,导致活性较低.本文采用浸渍法制备催化剂,以切开端口的短碳纳米管为载体,双重保证活性金属进入碳纳米管孔腔;引入稀土元素Ce抑制Cu的烧结,促进活性金属的均匀和分散,进而在保持CuCo催化剂的高活性的前提下使碳纳米管的限域效应在催化剂中得以发挥,使醇产物分布更为集中.
表1是本文所制的不同Ce含量的Ce-Cu-Co/CNTs催化剂催化合成气制低碳醇的活性数据.从表1中可以看出,获得的醇产物为C1-C4醇,同时在所有催化剂的醇产物中,乙醇所占比例都达到了42%以上,最高时可达51.1%.当Ce添加量为3%时,催化剂性能达到最佳,CO转化率、总醇时空收率和选择性分别为46.1%、696.4 mg∙g-1∙h-1和59.7%;乙醇选择性为27.9%,占总醇的46.8%;C3、C4醇选择性最低.
3.2 XRD表征
图1为不同Ce含量的Cu-Co基催化剂反应前、后的XRD图谱.由图可以看出,四种催化剂反应前后都检测到了2θ=26.1°处源于载体CNTs的类石墨态(002)面的衍射峰.反应前的催化剂中可以观察到明显的Cu2O的衍射峰和微弱的CuO的衍射峰,说明CuO呈高度分散状态.当Ce含量为3%时,Cu2O的衍射峰变得弥散,表明适量Ce的添加能显著增加Cu2O的分散度;当Ce的含量进一步增加至5%时,催化剂上可以观测到明显的CeO2衍射峰,同时Cu2O的衍射峰又变得尖锐.显然,过量的Ce会导致聚集态CeO2的形成,反而不利于Cu2O的分散.表2为催化剂反应前后通过半峰宽用谢乐方程(Sheerer equation)计算的Cu物种的平均晶粒直径.表2显示,当Ce含量由0%增加到3%,Cu物种的平均粒径由20.4 nm减小至16.5 nm;当Ce含量为5%时,Cu物种粒径又增大至21.2 nm.由图1(B)可以看出,催化剂的物相结构在反应后发生了明显的改变,均出现了Cu0的衍射峰,与反应前相同,Cu0晶粒度随Ce含量的增加先减小后增大,在Ce含量达到3%时达到最小.所有催化剂无论是反应前还是反应后均未观测到Co物种的衍射峰,表明Co呈无定形态或高分散态.此外,CuKα辐射源往往导致Co的XRD衍射图谱的背景噪音强,也使Co的衍射峰不明显.21

表1 催化剂上CO加氢反应性能Table 1 Catalytic performance of CO hydrogenation over catalysts

图1 不同Ce含量催化剂反应前(A)后(B)的XRD谱图Fig.1 XRD patterns of catalysts with different Ce contents before(A)and after(B)reaction
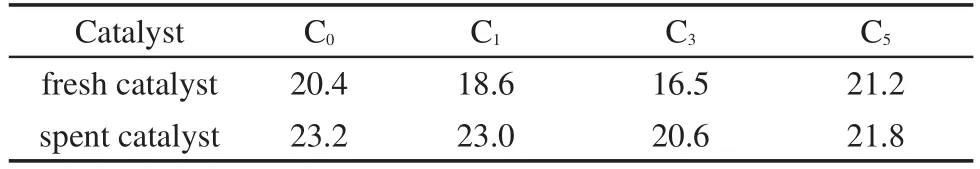
表2 不同Ce含量的催化剂反应前后Cu物种的平均晶粒度(nm)Table 2 Average particle size(nm)of Cu species of catalysts with different Ce contents before and after reaction
3.3 H2-TPR表征
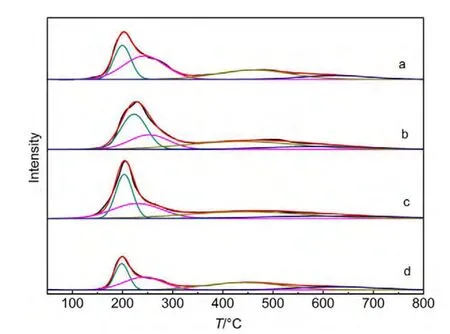
图2 不同Ce含量催化剂的H2-TPR谱图Fig.2 H2-TPR profiles of catalysts with different Ce contents
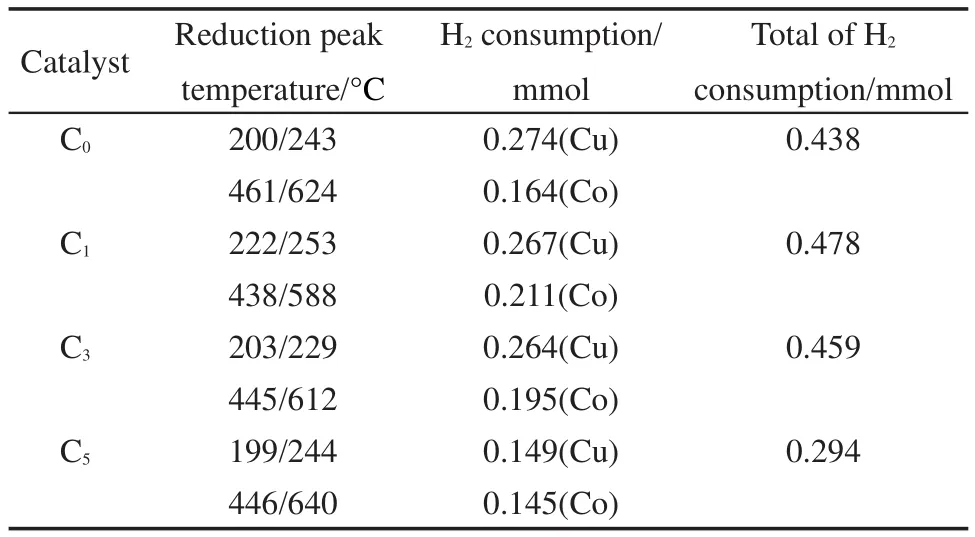
表3 H2-TPR计算的耗氢量Table 3 Hydrogen consumption from H2-TPR
图2显示的是不同Ce含量的CuCo基催化剂的H2-TPR图谱,对应的H2脱附峰耗氢量见表3.从图2可以看出,所有催化剂在200°C附近的主还原峰后都存在一个肩峰.在通常情况下,CuO直接还原为金属Cu而不经过中间态,并且CuO的还原要比Cu2O更加容易.22,23因此,结合前边的XRD表征和TPR的实验结果,认为200°C附近的主还原峰对应于CuO到Cu0的还原,后边的肩峰对应于Cu2O到Cu0的还原.此外,四个催化剂在350-800°C还出现有一个很宽的还原峰,由于纯的CeO2通常很难还原为单质Ce,只在800°C左右才能还原为Ce2O3,且CeO2与活性组分及载体间的相互作用使还原峰温进一步升高.24,25因此,本文将350-800°C的还原峰归属于Co3O4→CoO和CoO→Co0的两步还原的重合.26,27由表3可以看出,随着Ce含量的增加,催化剂耗氢量先增加后减小.当Ce含量增加至5%时,催化剂的耗氢量减小至0.294 mmol.结合XRD结果可以认为,在高Ce含量下,由于聚集态CeO2的形成,覆盖在Cu、Co物种表面,抑制了它们的还原.
3.4 BET和TEM表征
图3是不同Ce含量的催化剂N2吸/脱附曲线,相应的孔径分布见插图.由图3可以看出,含Ce和不含Ce的催化剂吸/脱附曲线和滞后环形状都没有明显变化,均属于IV型吸附等温线,且具有H1型迟滞环,是端口打开的碳纳米管类直通孔的表现.由孔径分布图可以看到,催化剂最可几孔径在2.5 nm左右,可归属于负载了活性金属后的碳纳米管的管径;10 nm以后的孔应为催化剂颗粒的堆积孔.
表4为本文所用碳纳米管和不同Ce含量催化剂的织构性质.从表4可以看出,单纯碳纳米管的比表面积是200.4 m2∙g-1,而催化剂的比表面积为170.0-186.8 m2∙g-1,相对于碳纳米管有所减少,这是活性金属沉积在管内壁所致.注意到相对于碳纳米管宽泛的管径(平均孔径15.48 nm,最可几孔径6.35 nm)而言,催化剂的孔径却极其集中(平均孔径9.49-9.98 nm,最可几孔径在2.58-2.77 nm),表明催化剂的活性组分不仅进入了碳纳米管内部,且充填进管内的金属量与碳纳米管管径可能存在密切关系.Li等28采用Monte Carlo模拟了Ni在碳纳米管中的添加,认为金属在大于0.76 nm直径的碳管内的充填,可以形成含有中心原子链和不含中心原子链的同心层状结构,且管径和原子排列层数值之间存在良好的线性关系.图4(a)为管径不同的两根碳纳米管重合在一起的TEM照片,可以明显看到管径不同,填充进管内的金属颗粒直径也不相同,且颗粒大小与管径相对应.由以上分析可以看出本文结果与这一结论相吻合,即金属原子在碳管内部形成了同心层状结构金属颗粒,且在不同管径的碳管填充的金属层数不同,从而使其宽泛的管径变得集中(见图4(b)中箭头所指).比较不加Ce的催化剂C0与加Ce的催化剂织构性质可以看出,当Ce加入量小于3%时,催化剂比表面积、孔容和平均孔径是略有增加的,整体变化不大;当加入量为5%时,上述参数均明显下降.结合前边表征可知,这是由于过量的Ce形成堆积,从而减小了催化剂的比表面积、孔容和平均孔径.

图3 不同Ce含量的催化剂N2吸脱附和孔径分布(插图)曲线Fig.3 N2-adsorption/desorption and pore size distribution(inset)curves of the catalysts with different Ce contents

表4 不同Ce含量催化剂的织构性质Table 4 Textural properties of catalysts with different Ce contents

图4 不同标尺下催化剂C3的TEM照片Fig.4 TEM pictures of C3with different scales
为了进一步考察催化剂的结构性质,挑选了具有代表性的C3进行了TEM表征.图4给出了四种不同标尺下的TEM图.图4显示,尽管催化剂样品在TEM测试时有些团聚,29但仍可以明显看出活性组分主要负载在碳纳米管内部.Santiso等30的理论计算表明,当化学反应限制在尺寸很小的管径内时,反应动力学明显改变,反应速率大幅提高.Bao等15将Rh-Mn纳米粒子组装到碳管内部,发现能够显著提高催化剂还原性能,促进CO的吸附和解离,明显提高了C2含氧化合物的产率.显然,本文催化剂很好地结合了CuCo催化剂的高活性和碳纳米管的限域效应,使催化剂活性和乙醇选择性均达到较优异的效果.
3.5 CO-TPD表征
在合成气制低碳醇反应中,要使反应能够有效地进行,必须使CO和H2能够在催化剂表面进行充分的活化.低碳醇中烃基的形成要求C―O键的断裂,而醇羟基的生成需要C―O键的保留,因此通过CO-TPD研究CO在催化剂表面的吸、脱附性质是探究催化剂催化合成低碳醇机理的重要手段.图5为不同Ce含量的催化剂反应前的CO-TPD谱图.由图可知,Ce的添加并没有改变催化剂上CO吸附中心种类,所有催化剂均存在3个CO脱附峰:100°C以下的低温脱附峰,400-500°C之间的中温脱附峰和600°C以后的高温脱附峰.Ce的引入对低、高温CO脱附峰的影响不大,但是对中温脱附峰有明显的影响.Ce的加入明显增加了CO中温脱附峰的强度:当Ce含量为1%和3%时增加幅度明显,到5%时又迅速下降,与催化剂活性有较好的对应关系,表明低碳醇的生成与中强吸附的CO密切相关.
一般认为,形成低碳醇的反应机理符合Xu等20提出的模型,即包括CO的解离吸附,C物种的加氢生成CHx,催化剂表面非解离吸附的CO插入CHx,最后再进行加氢生成低碳醇.通常强吸附导致CO的解离,而中强吸附导致C―O键保留的非解离吸附.18前者导致烃链的增长,后者导致含氧化合物的形成,因此CO的插入代表链的终止,1,31这与COTPD表征结果一致,即适量Ce的加入显著增加了催化剂表面较强吸附(非解离吸附)CO的活性位,提高CO物种的活化吸附量,从而更有利于低碳醇的形成;同时碳纳米管的限域效应由于限制了活性金属粒子的尺寸而使CHx的链增长受到了抑制.可见,本文催化体系的醇生成机理与Xu等20提出的机理相吻合,获得的性能特征符合纳米管的限域效应.15此外,注意到从醇分布看,Ce的加入量与甲醇的选择性有顺变趋势,与乙醇选择性有逆变关系,但总体上C2以上醇变化不显著,表明碳链增长与Ce的助催化作用不密切相关.这进一步证实了链增长来自于活性金属表面的CHx偶联,本文催化剂产物分布窄是碳纳米管限域效应的结果.
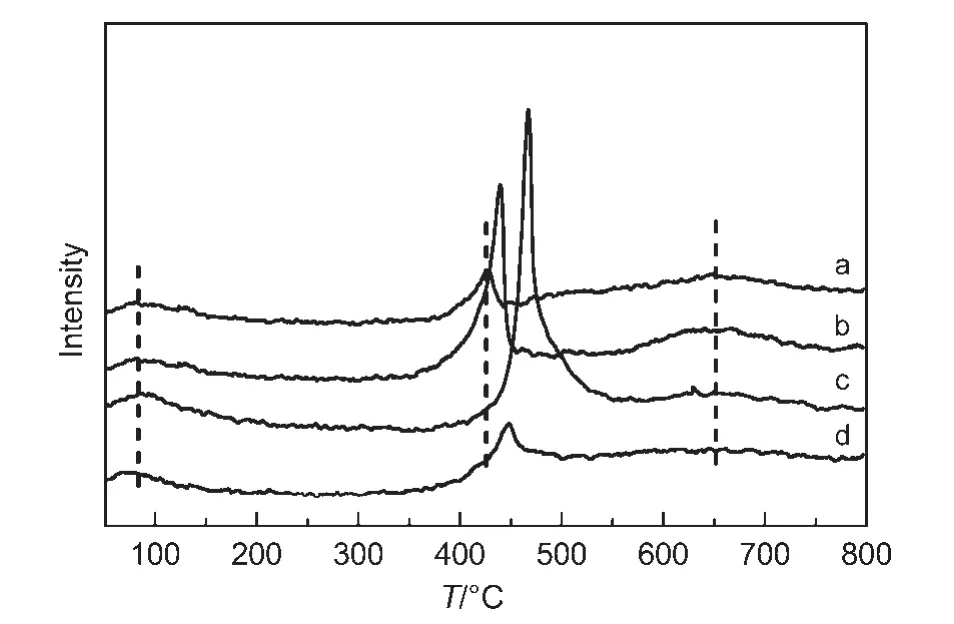
图5 不同Ce含量催化剂的CO-TPD谱图Fig.5 CO-TPD profiles of catalysts with different Ce contents
4 结论
本文研究表明,采用传统浸渍法以开口短碳纳米管为载体,可以制备出具有明显限域效应的高活性、高醇选择性的CuCo/CNTs基低碳醇合成催化剂,低碳醇的时空收率和选择性最高可达696.4 mg∙g-1∙h-1和59.7%,其中乙醇占总醇的46.8%,相关性能指标优于当前文献报道;适量Ce的引入明显改善了CO的非解离吸附、大幅度提高了催化剂活性和醇选择性,但与碳链的增长不密切相关;碳纳米管的限域效应由于限制了活性金属粒子的尺寸而使CHx的链增长受到了抑制,是本文催化剂产物碳数分布窄、乙醇选择性高的原因.
(1)Li,D.B.;Ma,Y.G.;Qi,H.J.;Li,W.H.;Sun,Y.H.;Zhong,B.Prog.Chem.2004,16,584.[李德宝,马玉刚,齐会杰,李文怀,孙予罕,钟 炳.化学进展,2004,16,584.]
(2)Ran,H.F.;Fang,K.G.;Lin,M.G.;Sun,Y.H.Nat.Gas Chem.Ind.2010,35,1.[冉宏峰,房克功,林明桂,孙予罕.天然气化工,2010,35,1.]
(3)Xiao,K.;Bao,Z.H.;Qi,X.Z.;Wang,X.X.;Zhong,L.S.;Fang,K.G.;Lin,M.G.;Sun,Y.H.Chin.J.Catal.2013,34,116.
(4)Zheng,W.;Yao,X.G.;Hu,D.Z.;Wen,L.Q.;Wang,X.Q.Sci.Technol.Inf.2009,10.[郑 伟,姚喜贵,胡大志,文良起,王小倩.科技资讯,2009,10.]
(5) Shi,L.M.;Chu,W.J.Mol.Catal.(China)2011,25,316.[士丽敏,储 伟.分子催化,2011,25,316.]
(6) Lin,M.G.;Fang,K.G.;Li,D.B.;Sun,Y.H.;Chin.J.Catal.2008,29,559.[林明桂,房克功,李德宝,孙予罕.催化学报,2008,29,559.]
(7)Wang,N.;Fang,K.G.;Lin,M.G.;Jiang,D.;Li,D.B.;Sun,Y.H.Nat.Gas Chem.Ind.2010,35,6.[王 宁,房克功,林明桂,姜 东,李德宝,孙予罕.天然气化工,2010,35,6.]
(8) Chen,X.P.;Zhao,N.;Sun,Y.H.;Ren,J.;Wang,X.Z.;Zhong,B.Coal Convers.1998,21,22.[陈小平,赵 宁,孙予罕,任 杰,王秀芝,钟 炳.煤炭转化,1998,21,22.]
(9) Shi,L.M.;Chu,W.;Deng,S.Y.J.Fuel Chem.Technol.2012,40,436.[士丽敏,储 伟,邓思玉.燃料化学学报,2012,40,436.]
(10) Pan,H.;Bai,F.H.;Su,H.Q.Chem.Ind.Eng.Prog.2010,29,157.[潘 慧,白凤华,苏海全.化工进展,2010,29,157.]
(11) Mao,D.S.;Guo,S.Q.;Yu,J.;Han,L.P.;Lu,G.Z.Acta.Phys.-Chim.Sin.2011,27,2639.[毛东森,郭胜强,俞 俊,韩璐蓬,卢冠忠.物理化学学报,2011,27,2639.]doi:10.3866/PKU.WHXB20111125
(12) Huang,L.H.;Chu,W.;Hong,J.P.;Luo,S.Z.Chin.J.Catal.2006,27,596.[黄利宏,储 伟,洪景萍,罗仕忠.催化学报,2006,27,596.]doi:10.1016/S1872-2067(06)60033-8
(13)Dong,X.;Liang,X.L.;Li,H.Y.;Lin,G.D.;Zhang,P.;Zhang,H.B.Catal.Today2009,147,158.doi:10.1016/j.cattod.2008.11.025
(14)Wang,M.W.;Li,F.Y.;Peng,N.C.New Carbon Mater.2002,17,75.[王敏炜,李凤仪,彭年才.新型碳材料,2002,17,75.]
(15) Pan,X.L.;Fan,Z.L.;Chen,W.;Ding,Y.J.;Luo,H.Y.;Bao,X.H.Nat.Mater.2007,6,507.
(16)Shi,L.M.;Chu,W.;Xu,H.Y.;Deng,S.Y.Rare Metal Mater.Eng.2009,38,1382.[士丽敏,储 伟,徐慧远,邓思玉.稀有金属材料与工程,2009,38,1382.]
(17)Wang,J.J.;Chernavskii,P.A.;Wang,Y.;Khodakov,A.Y.Fuel2013,103,1111.doi:10.1016/j.fuel.2012.07.055
(18)Shi,L.M.;Chu,W.;Deng,S.Y.J.Nat.Gas Chem.2011,20,48.doi:10.1016/S1003-9953(10)60145-4
(19) Cai,Q.R.;Peng,S.Y.The Catalysis Action in C1 Chemistry.Chemical Industry Press:Beijing,1995;p137.[蔡启瑞,彭少逸.碳-化学中的催化作用.北京:化学工业出版社,1995:137.]
(20) Xiao,D.X.;Doesburg,E.S.J.Catal.Today1987,2,123.
(21) Nachal,D.S.;G.B.;Challa,S.S.R.K.;James,J.S.Catal.Today2009,147,100.doi:10.1016/j.cattod.2009.02.027
(22) Jae,Y.K.;Jose,A.R.;Jonathan,C.H.;Anatoly,I.F.;Peter,L.L.J.Am.Chem.Soc.2003,125,10684.doi:10.1021/ja0301673
(23) Christpher,S.P.;Hari,N.;Chelsey,D.B.J.Catal.2009,266,308.doi:10.1016/j.jcat.2009.06.021
(24) Jing,G.;Jian,Z.G.;Dan,L.;Zhao,Y.H;Jin,H.F.;Xiao,M.Z.Int.J.Hydrog.Energy2008,33,5493.doi:10.1016/j.ijhydene.2008.07.040
(25) Damyanva,S.;Bueno,J.M.C.Appl.Catal.A2003,253,135.doi:10.1016/S0926-860X(03)00500-3
(26) Shu,J.J.;Shao,Q.S.Appl.Catal.B2013,140-141,1.
(27) Fang,Y.Z.;Liu,Y.;Zhang,L.H.Appl.Catal.A2011,397,183.doi:10.1016/j.apcata.2011.02.032
(28) Li,H.Y.;Ren,X.B.;Guo,X.Y.Chem.Phys.Lett.2007,437,108.doi:10.1016/j.cplett.2007.02.015
(29) Shu,J.C.;Xiu,L.P.;Liang,Y.;Xin,H.B.Mater.Lett.2011,65,1522.doi:10.1016/j.matlet.2011.02.070
(30) Santiso,E.E.;Kostov,M.K.;George,A.M.;Nardelli,M.B.;Gubbins,K.E.Appl.Surf.Sci.2007,253,5570.doi:10.1016/j.apsusc.2006.12.121
(31) Guo,S.Q.;Mao,D.S.;Yu,J.;Han,P.L.J.Fuel Chem.Technol.2012,40,1103.[郭强胜,毛东森,俞 俊,韩璐蓬.燃料化学学报,2012,40,1103.]
猜你喜欢
潍坊学院学报(2021年2期)2021-07-22 07:59:04
山西化工(2019年4期)2019-02-17 09:36:46
西南石油大学学报(自然科学版)(2018年2期)2018-06-26 06:19:12
物理化学学报(2018年1期)2018-01-29 07:45:45
雷达学报(2017年1期)2017-05-17 04:48:53
光学精密工程(2016年1期)2016-11-07 09:01:53
浙江大学学报(工学版)(2016年9期)2016-06-05 09:20:52
分析测试学报(2015年8期)2016-01-13 06:19:35
浙江工商大学学报(2015年3期)2016-01-07 06:51:07
原子与分子物理学报(2015年3期)2015-11-24 12:49:39
