
NMF模型在挖掘基因功能关系中的研究与应用
2014-02-09 07:47孙泽强谢红薇郝晓燕
计算机工程与设计 2014年4期
孙泽强,谢红薇,郝晓燕
(太原理工大学计算机科学与技术学院,山西太原030024)
0 引 言
随着生物医学文献的快速增长和学科界限的打破,跟踪这些新的发现就成了一个很大的挑战。此外,有关染色体组和蛋白质组的技术的最新发展,以及大量染色体组信息涌入生物医学研究,使得我们很难判断基因之间复杂的功能关系。由于这方面的研究既费时又昂贵,所以尽可能地从现有的文献中挖掘信息变得越来越重要。为了帮助研究人员利用现有的生物医学文献和基因组信息,已经有大量的人力物力投入到了开发有效的数据挖掘工具中。
Semantic gene organizer(SGO)是一种最有效的工具之一,使用潜在语义索引(LSI),执行截断奇异值分解(SVD),提取和筛选基因之间的功能关系[1]。Xu lijing等人验证了使用LSI从生物医学文献中提取明确的(直接)和隐藏的(间接)基因之间的功能关系的有效性。基本的SVD分解技术是将一个术语-基因文档矩阵分解为一系列新的因子矩阵,在低维子空间中,这些因子矩阵既可以用来描述术语,也可以用来描述文档。然而,很难直观地解释LSI的因子。LSI可以确定哪些基因是相关的,却很难说明这些基因为什么相关。
Li Fang使用非负矩阵分解(NMF)的方法,保持了原始数据矩阵的非负性[2]。NMF产生的低阶因子矩阵也被解释为数据的一部分,这种特性在科学、工程和医学等许多领域得到应用[3]。最近,NMF被广泛地应用在生物信息学领域中的,包括基因表达数据的分析,序列的分析,基因列表的功能特性和文本挖掘[4-9]。Kang等人已经证实了NMF方法在生物医学文献的语义特征提取中的有效性[7]。
于是,我们将NMF方法应用到挖掘文献中复杂的基因之间的功能关系,这样不仅可以确定哪些基因是相关的,还能巧妙地利用NMF产生的低阶因子矩阵,解释为什么这些基因相关,以及表示它们之间的相关程度。
1 GFRA的设计思路
在这项研究中,我们开发了一个基于Web的生物信息学软件环境,叫做基因功能关系助手(GFRA),便于发现基因之间的功能关系并根据基因之间的相关程度将其分类。GFRA可以挖掘出术语-基因文档数据的非负矩阵,并使用NMF方法提取出不同术语-基因文档数据中相关词的概念特征值。简单地说,给定一个m×n维非负矩阵A和一个整数k,满足0<k<min(m,n),我们要找到一个m×k维非负矩阵W和一个k×n维非负矩阵H,使得下列函数取得最小值:这样就把这个问题转化为可计算的问题,我们使用迭代法,不仅可以产生良好的数学近似模型,还能提供宝贵的生物信息。
GFRA的工作流设计如图1所示。由GFRA用户提供一个基因列表,使用和SGO同样的方法创建一个术语-基因文档矩阵,基于这个术语-基因文档矩阵建立NMF模型。简单地说,在Entrez Gene中查找和基因表中的所有基因相关的题目和摘要。目前,为了避免多义术语(一个术语有多重含义)和同义术语(多个术语的含义相同)的问题,这些摘要必须经过人工删除含义不明确的术语。将来要实现自动化处理摘要数据。将与一个特定基因相关的标题和摘要连接在一个基因文档中,用当前C++版本的通用文本解析器(GTP)将基因文档的集合解析为术语[10]。用非负的权值来表示一个术语和相应基因文档的关系,这些权值作为矩阵的元素,就形成了术语-基因文档矩阵。通过给特定的术语赋更大的权值,可以降低噪声术语的影响[11]。使用NMF方法分解术语-基因文档矩阵[12],将原始矩阵分解为因子矩阵,生成基因文档集合相应的k阶NMF模型。目前,GFRA分别取k的值为10、15和20,表示低、中、高解析率的NMF模型。以后,我们将根据用户的反馈确定k的合适的取值范围。用NMF模型提取与k个特征值对应的主术语和主基因,将主基因和每个特征值建立联系,GFRA用户对特征值添加注释。GFRA分类器就可以用带注释的NMF模型来确定新基因文档的概念特征值。
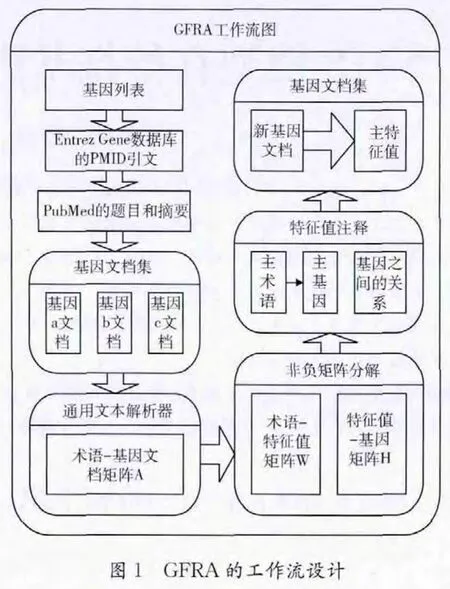
2 GFRA的工作过程
GFRA生物信息学软件环境的基础框架已建成,描述如下:
2.1 提取概念特征值
由于NMF模型具有非负性,所以可以从术语-特征值矩阵(W)中提取出概念特征值。稀疏矩阵的每一列(特征值)表示一个术语子集,从而形成了一种确定的术语使用模式。这种模式可以帮助GFRA用户确定特征值对应的概念。例如,一个包含了蚊子,疟原虫,血液,和奎宁的术语的特征值可能描述的是一种叫疟疾的疾病。一旦用户根据主术语识别出特定的特征值,这个特征值就可以解释成更具实际意义的术语(比如乳腺癌)而不是默认的标签(特征值5),以备将来参考。图2中是一些概念特征值和50TG的基因文档集(数据集1)中与这些特征值最相关的术语。用户可以用滤熵器来了解术语在整个过程中是如何使用的。如果一个术语以同样的方式出现在所有文件中,那么它很可能不是一个特别好的模型指标,所以赋给它一个低的熵权。高熵权的术语更明确,更有实际意义。这个滤熵器选项可以帮助用户选出更重要的特征值。
2.2 发掘关联基因
我们可以从特征值-基因矩阵(H)中提取出与每个特征值最相关的所有基因,同时,多个特征值也可以描述同一个基因。H矩阵的元素决定了基因和特征值的关系强度。有相同特征值的不同基因可能在功能上有一定的关联。图3列出了与特征值2最相关的基因和50TG的基因文档集中最相关的术语。
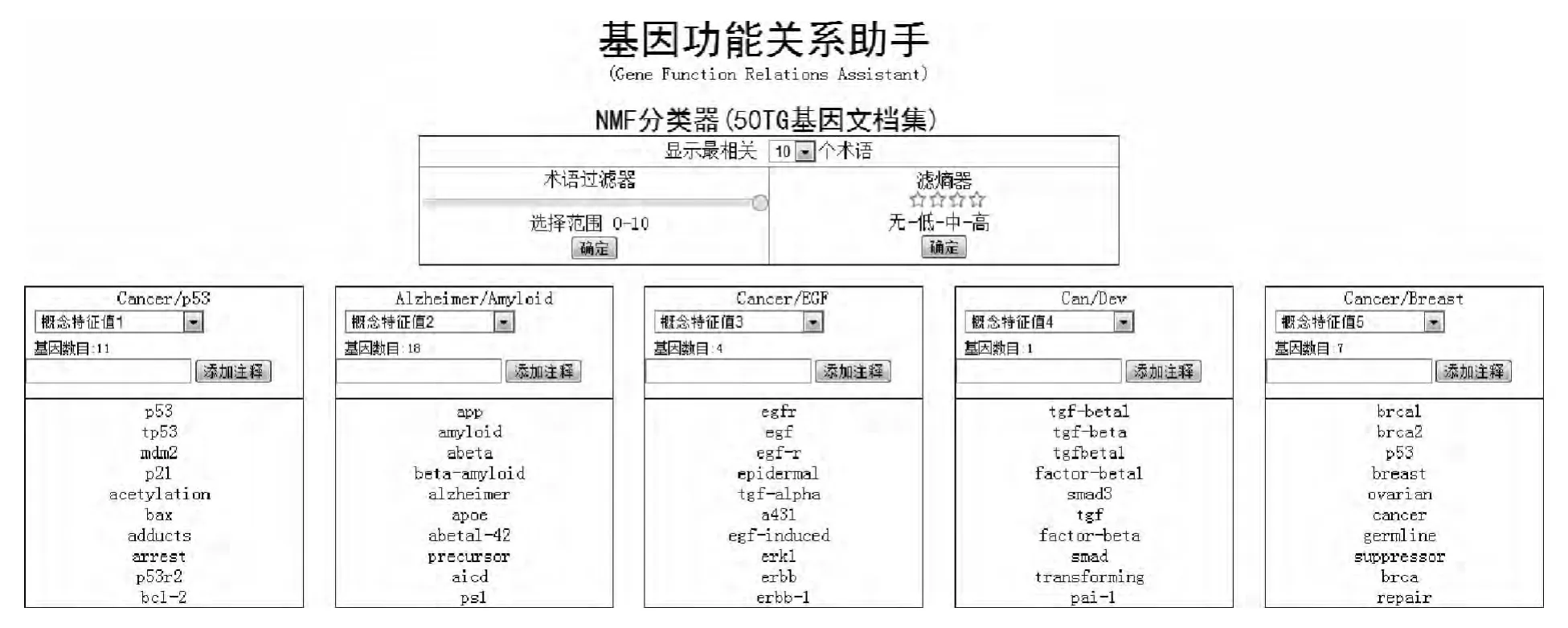
图2 概念特征值和与之最相关的术语
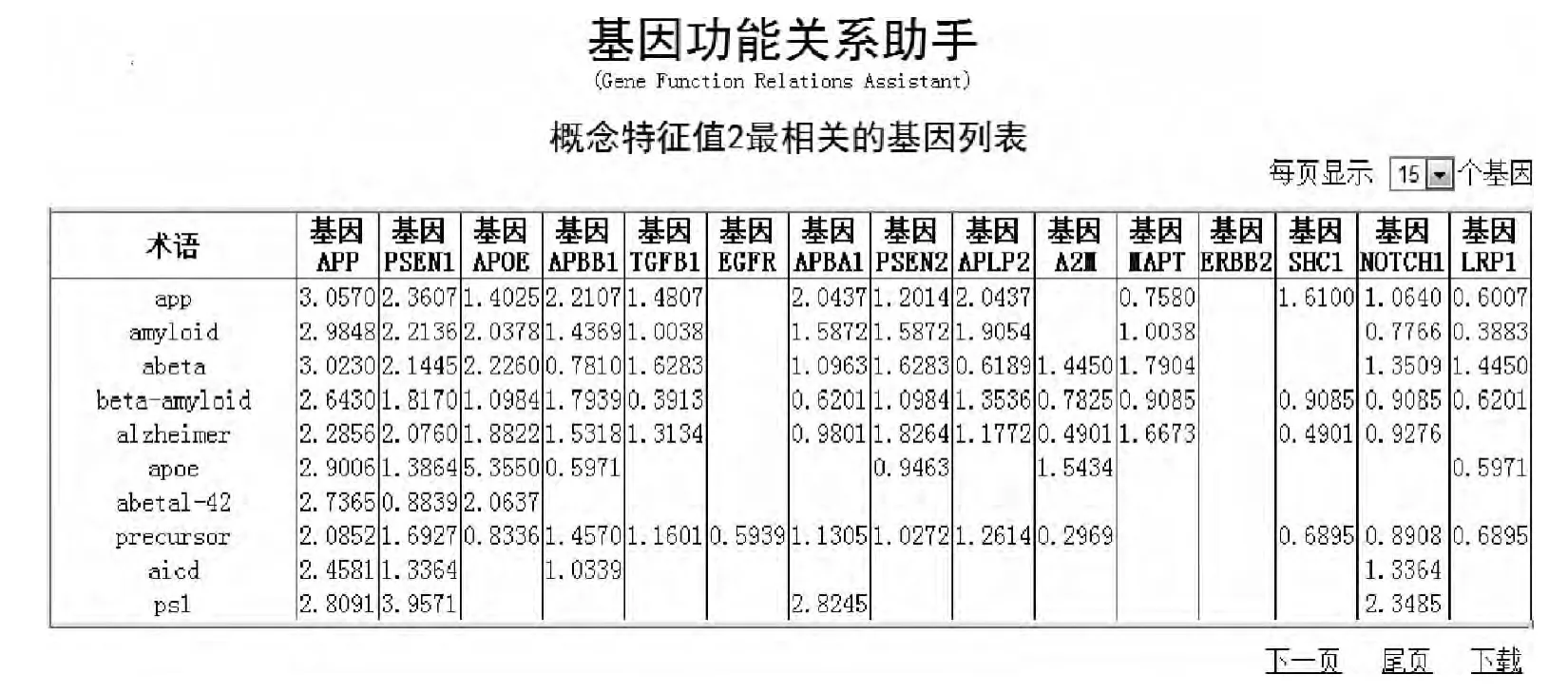
图3 与特征值2最相关的基因和术语列表
2.3 探寻基因之间的关系
上述内容描述了GFRA识别基因子集的过程。为了明确对于用户选择的特征值i,基因之间的关系强度,我们使用Pearson关系系数r来估计基因x和基因y的关系对于所有被用户选择的n个特征值,r的计算方法如下所示
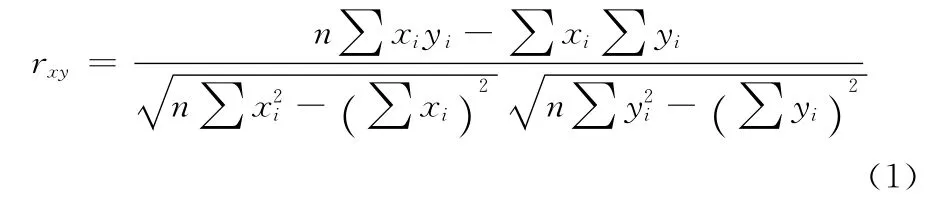
这样就生成了一个所有基因的Pearson关系矩阵。为了便于可视化分析,我们对相互关系进行了颜色编码,即关系越强,红色越深。用户可以选择3个以上的特征值组合查看,系统默认选择和用户所选的特征值相邻的左右两个特征值。选择前5个特征值组合查看,生成的50TG的基因文档集中与特征值2相关的所有基因的关系矩阵如图4所示。
GFRA的另外一个重要的功能是解释为什么一个基因的子集是相关的。两对基因对关于某个特征值有不同的关系强度,但是从整体上看,这两对基因对可能有类似的关系强度。用户可以点击关联单元格查看基因对关于所有特征值的关系强度。
2.4 给新的基因文档分类
新加入基因文档集的基因文档可以直接进行分析,而无需更新NMF模型。将新的文档数据流输入GFRA分类器,根据带注释的特征值,就可以确定这些新的基因文档对应的特征值。
3 实验结果的评价分析
我们用已知功能关系的基因,手动构建了3个基因文档数据集,用于初步评估GFRA的特征值分类器。第一个数据集(50TG)是一个由50个手动选择的基因组成的基因文档集合,这些基因与细胞生长,阿尔茨海默氏症,癌症生物学相关[1]。第二个数据集是从Biocarta,Gene Ontology和MeSH数据库中抽取出的3个不重叠的基因列表。第三个数据集是从Nature Reviews文章中选出的5个基因列表。表1是这3个数据集的分类情况。

图4 与特征值2相关的所有基因的关系矩阵
对于每个数据集,分别取k=10,15,20,30,40和50,生成6个NMF模型。根据特征值的主术语,为NMF模型中的特征值手动添加注释,并分为一个或者多个类别。然后,根据这些已经注释的特征值,将数据集中的基因分配到与其关系强度最大(矩阵H的列元素最大)的类别中。例如,如果基因x在矩阵H中对应的列元素的最大值对应的特征值是i,那么说明基因x与特征值i的关系强度最大,就把基因x分配到特征值i所标识的类别中。GFRA分类器用最强特征值法分别对数据集1,2和3中的基因进行了分类,分类准确率如图5所示,横坐标表示不同阶数k对应的NMF模型,纵坐标表示每种NMF模型对不同数据集分类得到的准确率,取k=30,得到的准确率分别为98%,88.2%和86.4%。从图5可以看出,当阶数k=10时,分类器的准确率比较低,随着k值的增加,准确率逐渐提升,当阶数k大于等于30时,准确率基本稳定,并保持较高水平。实验结果说明选取足够大的适当的阶数,可以达到预期的分类效果。
我们还用多特征值法分别对数据集1,2和3中的基因进行了分类。首先,找出每个特征值在矩阵H中的最大行元素(max H)对应的基因,设定一个阈值范围H>(max H)×系数q(其中0<q≤1),仅保留该行元素值在这个范围内的对应的其它基因。然后,对于每个基因,取所有与这个基因相关联并且在阈值范围内的所有特征值,然后根据这些特征值的标识,将这个基因归类。由于一个基因可能被分到几个不同类别中,所以我们用一个模糊方式对分类的准确率进行评估。如果分配给一个基因x的类别不包含正确的类别,那么基因x的分类正确性Ax为0。如果分配给一个基因x的类别包含正确的类别,那么基因x的分类正确性Ax为1/s,s表示分配给基因x的类别数。用t表示基因数目,总准确率A的计算方法如下所示


表1 数据集的分类情况
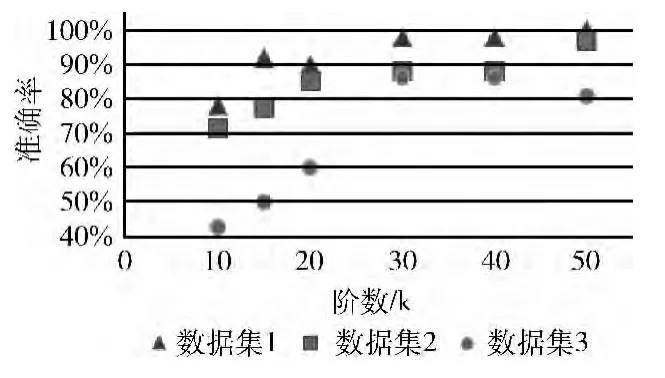
图5 GFRA分类器用最强特征值法得到的准确率
图6所示的是GFRA分类器用多特征值法得到的准确率,横坐标表示不同阶数k对应的NMF模型,纵坐标表示每种NMF模型采用不同系数值得到的分类准确率。取系数q为1.0,数据集1,2和3的准确率分别为78%-100%,71.6%-97.1%和42.7%-80.9%。整体上看,阶数相同的NMF模型随着系数值的增加,准确率逐渐提升。实验结果说明设定的阈值范围越高,分类效果越好。
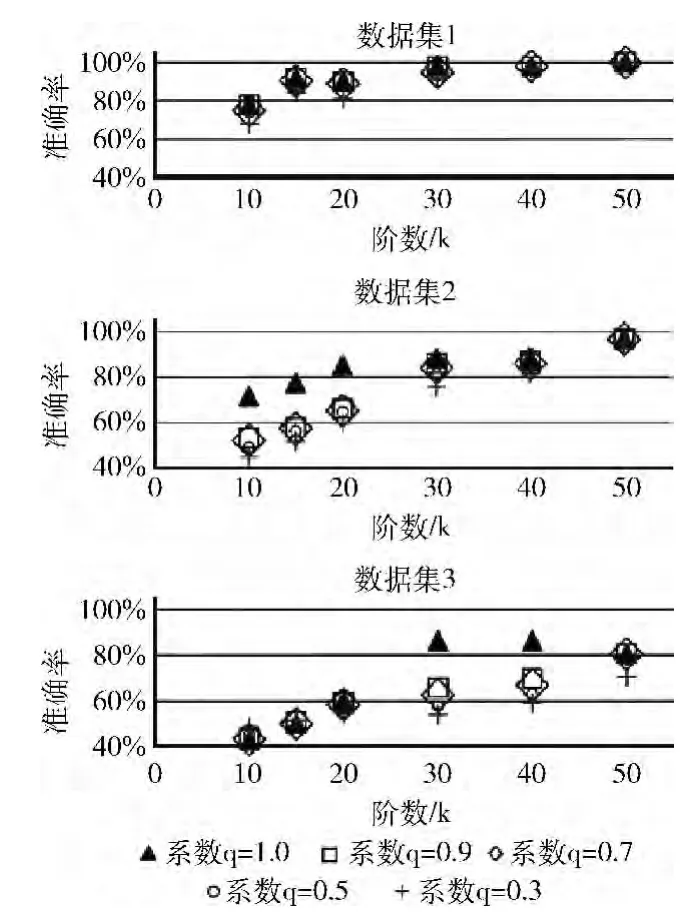
图6 GFRA分类器用多特征值法得到的准确率
4 结束语
给定一个基因列表,研究人员能够用GFRA来确定哪些基因是相关的,在满足一定精度要求的情况下,按功能将这些基因分类。我们已经分析了NMF模型的秩对分类结果的影响,考虑生成一个控制术语表,用于GFRA的分类功能,以满足相应目标函数的平滑性约束,保留熵值比较高的术语。GFRA不仅可以帮助研究人员有效的使用生物医学文献,而且还有知识发现的功能。
[1]Xu L,Furlotte N,Lin Y,et al.Functional cohesion of gene sets determined by latent semantic indexing of Pub Med abstracts[J].PloS One,2011,6(4):e18851.
[2]Li F,Zhu Q,Lin X.Topic discovery in research literature based on non-negative matrix factorization and testor theory[C]//Information Processing Asia-Pacific Conference on,2009:266-269.
[3]Jeon K M,Park N I,Kim H K,et al.Mechanical noise suppression based on non-negative matrix factorization and multi-band spectral subtraction for digital cameras[J].IEEE Transactions on Consumer Electronics,2013,59(2):296-302.
[4]ZHANG Zhongyuan,ZHANG Xiangsun.NMF-based method for data classification[J].Computer Engineering and Applications,2010,46(16):245-248(in Chinese).[张忠元,章祥荪.NMF的数据分类方法在肿瘤分类上的应用[J].计算机工程与应用,2010,46(16):245-248.]
[5]TAO Weijie,KONG Wei.Discovering gene expression regulatory networks of incipient AD based on NMF[J].Journal of Anhui University:Natural Science Edition,2012,36(1):69-75(in Chinese).[陶伟杰,孔薇.基于NMF技术探寻早期AD基因表达调控网络[J].安徽大学学报(自然科学版),2012,36(1):69-75.]
[6]CAO Shengyu,LIU Laifu.Non-negative matrix factorization and its applications to gene expression data analysis[J].Journal of Beijing Normal University:Natural Science,2007,43(1):30-33(in Chinese).[曹胜玉,刘来福.非负矩阵分解及其在基因表达数据分析中的应用[J].北京师范大学学报(自然科学版),2007,43(1):30-33.]
[7]Kang B C,Sur Z W,Park C,et al.Document clustering of MEDLINE abstracts based on non-negative matrix factorization using local confidence assessment[J].BioChip Journal,2010,4(4):336-349.
[8]GAO Maoting,WANG Zheng'ou.Comparing dimension reduction methods of text feature matrix[J].Computer Engineering and Applications,2006(30):157-159(in Chinese).[高茂庭,王正鸥.几种文本特征降维方法的比较分析[J].计算机工程与应用,2006(30):157-159.]
[9]SHI Jinlong,LUO Zhigang.Research on the advances of nonnegative matrix factorization and its application in bioinformatics[J].Computer Engineering and Science,2010,32(8):117-123(in Chinese).[石金龙,骆志刚.非负矩阵分解算法及其在生物信息学中的应用研究[J].计算机工程与科学,2010,32(8):117-123.]
[10]Manine,A P,Alphonse E,Bessieres P.Learning ontological rules to extract multiple relations of genic interactions from text[J].International Journal of Medical Informatics,2009,78(12):31-38.
[11]Wang Y X,Zhang Y J.Image inpainting via weighted sparse non-negative matrix factorization[C]//18th IEEE International Conference on Image Processing,2011:3409-3412.
[12]Heinrich K E,Berry M W,Homayouni R.Gene tree labeling using nonnegative matrix factorization on biomedical literature[J].Computational Intelligence and Neuroscience,2008(2008):12.
猜你喜欢
电子产品世界(2022年4期)2022-04-21
苏州市职业大学学报(2021年1期)2021-04-08
烟台大学学报(自然科学与工程版)(2021年1期)2021-03-19
计算机系统应用(2021年2期)2021-02-23
计算机应用与软件(2020年1期)2020-01-14
中国外汇(2019年13期)2019-10-10
计算机测量与控制(2019年4期)2019-05-08
课程教育研究·新教师教学(2016年18期)2017-04-12
科技经济市场(2014年11期)2014-12-30
