
17β-雌二醇介导的信号通路对急性呼吸窘迫综合征小鼠肺泡上皮钠离子通道的调控研究
2014-02-08 07:11冯龙华王导新
中国全科医学 2014年27期
戚 迪,何 婧,叶 媛,冯龙华,王导新
非心源性肺水肿造成的氧合功能障碍是导致急性呼吸窘迫综合征(ARDS)顽固性低氧血症的特异性原因,因此,如何维持有效肺泡水肿液清除(alveolar fluid clearance,AFC)是改善气体交换、提高血氧饱和度、降低病死率的关键[1-2]。肺泡上皮钠通道(epithelial sodium channel,ENaC)是由α、β、γ亚基组成三聚体,作为肺泡上皮钠水转运的限速环节,在ARDS所致肺水肿的发生发展中扮演重要的角色,其调控机制及通路的研究具有重要价值[3-4]。多项研究表明,17β-雌二醇作为一种重要的类固醇激素可通过多方面作用改善ARDS[5-10]。β-雌二醇对于ENaC的经典调节机制是通过17β-雌二醇受体反应元件(estrogen relative element,ERE)介导的基因转录调节,但近年来,其通过快速激活细胞内信号通路介导的非基因转录后调节机制引起关注[11-12]。众所周知ENaC的活化及表达受AGC激酶家族(protein kinase A/protein kinase G/protein kinase C)的调节,其中SGK1(serum-glucocorticoid-induced kinase 1)作为一种重要的AGC激酶,在调控离子液体转运中扮演重要角色[13-15]。磷脂酰肌醇3-激酶(phosphoinositide 3-kinase,PI3K)及其直接靶蛋白——蛋白激酶B(protein kinase B,PKB/AKT) 是机体应对损伤的一条重要的内源性负反馈通路,且与ARDS发生发展关系密切[16-19]。但17β-雌二醇通过该通路对ENaC的调节作用研究甚少。本研究旨在通过建立ARDS小鼠模型,观察17β-雌二醇介导的PI3K/AKT/SGK1信号通路对ARDS ENaC的调控作用及机制。
1 材料与方法
1.1 实验动物 SPF级雄性C57B/L6小鼠40只,5~8周龄,体质量(20±5) g〔重庆医科大学动物实验中心提供,许可证号:SCXK(渝) 2012-0001〕,普通光照环境下饲养,自由饮水与摄食喂养。
1.2 实验材料 脂多糖(LPS,E.coli,O111:B4)、渥曼青霉素及17β-雌二醇购于Sigma公司;髓过氧化物酶(MPO) 检测试剂盒购于南京建成工程生物研究所;总RNA 提取试剂盒、cDNA合成试剂盒、反转录聚合酶链式反应(RT-PCR)试剂盒、DNA marker DL1000 购于宝生物工程(大连)有限公司;RIPA裂解液(RIPA Lysis Buffer)及BCA蛋白浓度测定试剂盒购于碧云天生物技术研究所;酶联免疫吸附试验(ELISA)试剂盒购于北京四正柏生物科技有限公司;β-肌动蛋白(β-actin)、α-、β-、γ-ENaC单克隆抗体购于Abcam 公司;磷酸化AKT(p-AKT,Ser473)抗体购于Cell Signaling 公司;磷酸化SGK1(p-SGK1,Ser422)多克隆抗体购于Epitomics公司;ECL检测试剂盒购于南京凯基生物科技发展有限公司。
1.3 实验方法
1.3.1 小鼠ARDS模型建立 40只小鼠采用随机数字表法分为对照组(control组)、模型组(LPS组)、17β-雌二醇治疗组(E2组)、渥曼青霉素干预组(wortmannin组),每组10只。腹腔注射4%水合氯醛(0.2 ml/20 g)麻醉小鼠。LPS组、E2组、wortmannin组通过气管插管注射5 mg/kg无菌LPS(10 μg LPS溶于50 μl无菌0.9%氯化钠溶液),诱导建立ARDS模型,并根据血气分析判断建模成功。control组给予无菌0.9%氯化钠溶液代替LPS。E2组及wortmannin组于LPS注射后20 min分别腹腔注射17β-雌二醇(1 mg/kg)。wortmannin组分别于 LPS 注射前90 min,注射后 90 min、360 min给予PI3K抑制剂渥曼青霉素(0.06 mg/kg),E2组给予等剂量0.9%氯化钠溶液。
1.3.2 苏木素-伊红(HE)染色观察小鼠肺组织病理变化并进行肺损伤评分 8 h处死小鼠,取右下肺组织,4%多聚甲醛固定,制成5 μm的石蜡切片,HE染色,光镜观察。肺损伤评分标准:(1)肺泡充血,(2)出血,(3)肺泡或血管腔内中性粒细胞浸润或聚集,(4)肺泡壁增厚和/或透明膜形成。依据病变轻重评0~4分(0分:无病变或非常轻微;1分:轻度病变;2分:中度病变;3分:重度病变;4分:极重度病变)。ARDS总评分为各项评定分数相加总和。
1.3.3 小鼠血气分析 根据实验分组,于LPS注射前30 min,注射后30 min 、1 h、4 h、8 h 经右侧颈总动脉采集1 ml动脉血,并用肝素抗凝(1∶1 000),立即送检,全自动血气分析仪检测动脉血氧分压(PaO2)。
1.3.4 小鼠肺水肿检测 造模8 h后处死小鼠取出肺组织,放置于事先称重的锡箔纸上称量肺湿重,再将样本置于80 ℃烤箱48 h至恒重后,称量其干重。根据公式计算肺湿干比(W/D=湿重/干重),反映肺水肿的严重程度。
1.3.5 小鼠肺泡灌洗液(BALF)检测 造模8 h后,以1 ml PBS分3次肺泡灌洗。收集前两次BALF,并于4 ℃ 14 000 r/min离心30 min(离心半径15 cm),取上清液,按照BCA蛋白测定试剂盒说明测定BALF总蛋白含量。按照ELISA试剂盒要求,BALF 4 ℃以400 r/min离心5 min(离心半径15 cm),沉渣以100 μl PBS液重悬,测定BALF中白介素(IL)-1β和肿瘤坏死因子α(TNF-α)水平。按照MPO试剂盒要求,检测BALF中MPO活性水平。
1.3.6 RT-PCR法检测小鼠肺组织α-、β-、γ-ENaC mRNA表达水平 采用RT-PCR 法,按照总RNA提取试剂盒说明书提取各组小鼠肺组织总RNA,紫外分光光度计测总RNA浓度。依据反转录试剂盒说明书,以小鼠肺组织总RNA为模板,反转录合成cDNA。冰上配制RT反应液如下:0.5 μg总RNA为模板,5×反转录缓冲液2 μl,Oligo(dT)18引物(50 μmol/L)0.5 μl,随机6核苷酸引物(100 mmol/L)0.5 μl,以DEPC水补充至10 μl。反应条件如下:37 ℃ 15 min,85 ℃ 5 s。按照扩增试剂盒说明书,建立PCR 反应体系如下:2 μl cDNA模板、Premix Taq 25 μl、正反引物各1 μl,以DEPC水补充至50 μl。引物序列如下:α-ENaC:上游引物5′-TACAACTCTTCCTACACTCGCCA-3′,下游引物5′-CTGGTTGAAACGACAGGTAAAGAT-3′;β-ENaC上游引物5′-CAATGACACCCAGTATAAGATGACC-3′,下游引物5′-CAATGAGGCACAGCACCGA-3′;γ-ENaC:上游引物5′-CAATGAGAACGAGAAGGGAAAG-3′,下游引物5′-AAGAAGCAGGTCACCAGCAGT-3′;内参β-actin:上游引物5′-CGAGCGGGCTACAGCTTC-3′,下游引物5′-GTCACGCACGATTCCCTCT-3′。反应条件如下:预变性 94 ℃ 5 min,变性94 ℃ 30 s,退火温度分别为:α-、β-ENaC 56 ℃;γ-ENaC、β-actin 57 ℃,退火时间分别为:α-ENaC 25 s;β-ENaC 17 s;γ-ENaC 18 s;β-actin 37 s。延伸72 ℃ 60 s,循环35轮,再延伸72 ℃ 10 min。PCR 产物10 μl与上样缓冲液混匀,2.5%琼脂糖凝胶电泳。凝胶成像,Quantity One软件进行分析,并以目的条带与β-actin 条带灰度的比值衡量α-、β-、γ-ENaC mRNA的转录水平。
1.3.7 Western blotting法检测小鼠肺组织α-、β-、γ- ENaC,p-AKT,p-SGK1蛋白的表达 按照试剂盒操作要求,RIPA裂解液提取肺组织总蛋白,BCA法测定蛋白浓度,-80 ℃ 保存。蛋白样品行聚丙烯酰胺凝胶电泳(SDS-PAGE),然后转移至硝酸纤维素滤膜(PVDF膜)上,5%脱脂奶粉或5%BSA封闭1 h,分别加入抗体α-ENaC(1∶300) 、β-ENaC(1∶1 000)、γ-ENaC(1∶1 000)、p-AKT(1∶1 000) 、p-SGK1(1∶500)、β-actin(1∶8 000)4 ℃ 孵育过夜。PBST洗膜3遍,10 min/次。加入HRP标记的二抗(1∶8 000),室温孵育1.5 h,PBST洗膜3遍,10 min/次。ECL显色。凝胶成像,并以Quantity One 软件分析目的条带与β-actin条带灰度的比值衡量α-、β-、γ-ENaC、p-AKT、p-SGK1蛋白的表达水平。
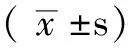
2 结果
2.1 HE染色观察小鼠肺组织病理变化并进行肺损伤评分 肺组织切片常规HE染色,光镜下观察。control组:肺组织未见肺泡腔内水肿出血或透明膜,肺泡间隔均一无增宽,未见炎性细胞浸润(见图1A)。LPS组:肺组织损伤明显,肺泡上皮肿胀严重,肺泡腔内及间质水肿出血严重。肺泡间隔增厚明显并伴大量炎性细胞浸润(见图1B)。E2组:肺组织损伤明显减轻,肺泡腔内水肿出血减轻,间隔轻度增宽,炎性细胞浸润减轻(见图1C)。wortmannin组:肺组织损伤较明显,肺泡及间质水肿出血较明显,肺泡间隔增厚伴炎性细胞浸润增加(见图1D)。LPS组肺损伤评分为(4.2±0.2)分,E2组为(2.7±0.5)分,wortmannin组为(3.9±0.3)分,差异有统计学意义(F=46.07,P=0.000);其中E2组较LPS组降低,差异有统计学意义(q=12.70,P=0.000),wortmannin组较E2组升高,差异有统计学意义(q=9.52,P=0.004)。
2.2 小鼠PaO2变化 4组不同时间PaO2比较,差异有统计学意义(F总=188.148,P总=0.000);其中LPS组、E2组、wortmannin组PaO2各个时间点较control组均下降,差异有统计学意义(P<0.05);E2组与LPS组比较,30 min、1 h时升高,差异有统计学意义(q=22.393,P=0.000;q=26.690,P=0.000),4 h、8 h时比较,差异无统计学意义(q=1.857,P=0.190;q=1.670,P=0.213)。wortamnin组与E2组比较,30 min、1 h时降低,差异有统计学意义(q=14.399,P=0.001;q=23.250,P=0.000),4 h、8 h时差异无统计学意义(q=0.415,P=0.527;q=0.379,P=0.546,见图2)。
2.3 小鼠肺组织W/D比较 4组小鼠肺组织W/D比较,差异有统计学意义(P<0.05);其中LPS组、E2组、wortmannin组较control组升高,LPS组、wortmannin组较E2组升高,差异均有统计学意义(P<0.05,见表1)。
2.4 小鼠BALF中总蛋白含量、IL-1β和TNF-α水平及MPO活性比较 4组小鼠BALF中总蛋白含量、IL-1β和TNF-α水平及MPO活性比较,差异有统计学意义(P<0.05);其中LPS组、E2组、wortmannin组以上指标较control组升高,LPS组、wortmannin组以上指标较E2组升高,差异均有统计学意义(P<0.05,见表1)。
2.5 各组小鼠肺组织α-、β-、γ-ENaC 的mRNA表达水平 RT-PCR法显示,4组小鼠肺组织α-、β-、γ-ENaC 的mRNA表达水平比较,差异均有统计学意义(F值分别为4.713、24.652、5.345,P值分别为0.007、0.000、0.004);其中LPS组、E2组、wortmannin组以上指标较control组降低,差异均有统计学意义(P<0.05);LPS组、E2组、wortmannin组比较,差异均无统计学意义(P>0.05,见图3)。
2.6 各组小鼠肺组织α-、β-、γ-ENaC表达 Western blotting法显示,4组小鼠肺组织α-、β-、γ-ENaC表达水平比较,差异均有统计学意义(F值分别为14.613、5.672、8.447,P值分别为0.000、0.003、0.000);其中LPS组和wortmannin组以上指标较control组和E2组下降,差异有统计学意义(P<0.05,见图4)。
2.7 各组小鼠肺组织p-AKT、p-SGK1表达水平 Western blotting法显示,4组小鼠肺组织p-AKT、p-SGK1表达水平比较,差异均有统计学意义(F值分别为8.371、8.862,P值分别为0.000、0.000);其中LPS组和wortmannin组以上指标较Control组和E2组下降,差异有统计学意义(P<0.05,见图5)。
注:PaO2=动脉血氧分压;与control组比较,#P<0.05;与E2组比较,*P<0.05;与wortmannin组比较,★P<0.05
图2 各组小鼠不同时间动脉PaO2变化
Figure2 Changes of PaO2in mice of each group

注:A control组,B LPS组,C E2组,D wortmannin组
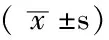
图1 小鼠肺组织病理改变(HE染色,×200)
注:W/D=肺湿干比,IL-1β=白介素1β,TNF-α=肿瘤坏死因子α,MPO=髓过氧化物酶;与control组比较,*P<0.05;与E2组比较,△P<0.05
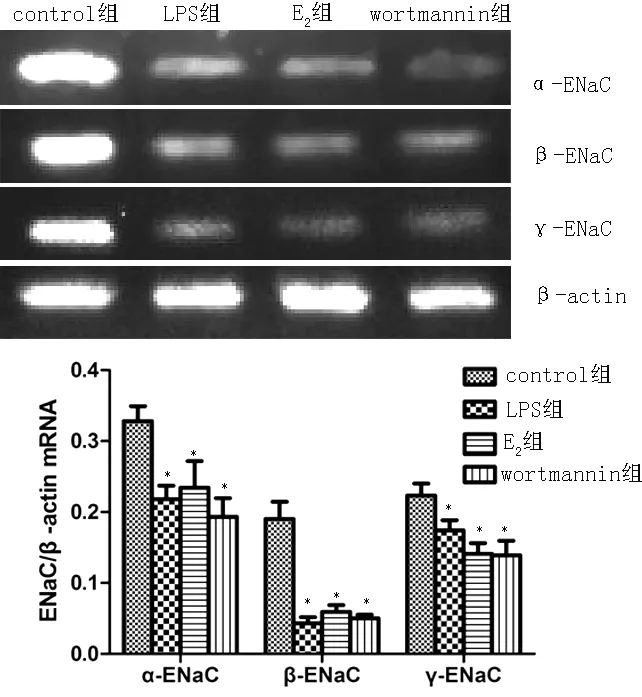
注:ENaC=肺泡上皮钠通道;与control组比较,*P<0.05
图3 各组小鼠肺组织α-、β-、γ-ENaC 的mRNA表达水平比较
Figure3 Comparison of α-,β-,γ-ENaC mRNA transcription in lung tissues of mice

注:与control组比较,#P<0.05;与E2组比较,*P<0.05
图4 各组小鼠肺组织α-、β-、γ-ENaC表达比较
Figure4 Comparison of α-,β-,γ-ENaC protein expression in lung tissue of mice

注:与control组比较,#P<0.05;与E2组比较,*P<0.05
图5 小鼠肺组织p-AKT、p-SGK1的蛋白表达
Figure5 p-AKT and p-SGK1 protein expression in lung tissue of mice
3 讨论
ARDS发病机制复杂,病死率高,主要的病理生理改变为弥漫性肺损伤,肺泡毛细血管内皮细胞与肺泡上皮细胞屏障的通透性增高,富含蛋白的炎性水肿液集聚于肺泡及肺间质,造成肺通气/血流比值失调,顺应性下降,临床表现为明显的呼吸窘迫、严重的顽固性低氧血症。非心源性肺水肿造成的氧合功能障碍是导致ARDS顽固性低氧血症的特异性原因[1]。因此,增强AFC对恢复患者肺换气能力、改善氧合具有积极意义[2]。ENaC作为钠离子跨膜转运的限速通道蛋白,协同肺泡基底膜的钠/钾ATP酶,在维持肺泡钠水转运平衡和促进AFC过程中发挥着主导作用[3]。实验发现α-、β-、γ- 3个亚基共同参与ENaC的组成及钠水转运功能,敲除或抑制某个亚基的表达都会导致Na+电流减小,造成肺泡钠水转运障碍,AFC显著下降,肺水肿加剧[4]。
目前,临床应用最有效的抗炎、消散肺水肿液药物糖皮质激素仍存在多种不良反应,17β-雌二醇作为机体内分泌系统产生的除糖皮质激素、盐皮质激素之外的另一种重要的类固醇激素,基础实验发现其可通过减轻炎性反应、抑制一氧化碳合酶、加速炎性细胞凋亡、上调水通道蛋白表达等多方面作用参与调节肺脏的结构及功能发育,与ARDS关系密切[5-8]。且临床研究发现相对于男性而言,女性对于严重创伤、脓毒血症、休克、溺水等因素诱发的ARDS具有更强的抵抗能力[9-10]。17β-雌二醇经典的作用途径是由17β-雌二醇受体(ER)与配体结合诱导ER与热休克蛋白(HSP)解离并形成E-ER复合物,直接或间接与17β-雌二醇反应原件(ERE)结合,从而正向或负向调节mRNA的转录,但近年来,越来越多的研究发现非基因性信号通路快速激活在17β-雌二醇保护作用中同样扮演着重要角色[11-12]。ENaC的表达及活化受多种AGC激酶的调节,如c-AMP/PKA、c-GMP/PKG、PKG通路。血清和糖皮质激素诱导的SGK1作一种重要的AGC激酶,在离子液体转运中扮演重要角色,其核心功能为通过抑制泛素连接酶nedd4-2介导的ENaC泛素化,增加ENaC在细胞膜上的表达量[13-15]。PI3K/AKT作为多种细胞信号通路和信号分子磷酸化级联反应的一个功能性交汇通路,在多种应激条件下可对机体发挥代偿性保护作用。本课题前期实验及国内外相关报道均证实该通路同样为介导盐皮质激素、糖皮质激素、胰岛素等对ENaC发挥正向调节作用的共用途径[16-19]。
因此推测17β-雌二醇可能通过快速激活PI3K/AKT/SGK1信号通路介导的非基因调控机制对ENaC发挥上调作用,从而增强AFC,减轻肺水肿,改善ARDS预后。本实验结果证实气管插管注射LPS造模后小鼠表现出明显的ARDS病理生理改变,17β-雌二醇干预后可产生快速抑制ARDS病理生理改变的短效作用,具体表现为:快速减轻肺水肿及炎性反应,降低W/D,降低BLAF中总蛋白含量及IL-1β、TNF-α、MPO活性水平,改善缺氧,提示17β-雌二醇对ARDS具有保护作用。本研究通过对ENaC蛋白、mRNA水平的测定证实:17β-雌二醇干预8 h后小鼠的ENaC亚基蛋白表达均有不同程度的增加,然而ENaC 亚基mRNA水平未明显改变,提示17β-雌二醇可能通过非基因转录后调节机制快速上调ENaC的蛋白表达,促进肺水肿液体吸收。实验结果进一步证实LPS组中p-AKT及p-SGK1水平降低,而E2组的p-AKT及p-SGK1水平增高,且17β-雌二醇对ENaC的正向调节作用可被PI3K的抑制剂渥曼青霉素阻止,进一步表明此上调作用至少部分是通过快速激活细胞相关信号通路PI3K/AKT/SGK1信号通路介导的基因转录后调控,但其具体分子调控机制及其与经典基因调控的协作关系还需进一步探讨。
综上所述,17β-雌二醇可通过PI3K/AKT/SGK1信号通路介导的基因转录后调节对ENaC 发挥正向调节作用,为推动ARDS病程中肺泡水肿清除,提高肺氧合能力提供基础理论根据。
1 Morty RE,Eickelberg O,Seeger W.Alveolar fluid clearance in acute lung injury:what have we learned from animal models and clinical studies? [J].Intensive Care Med,2007,33(7):1229-1240.
2 Lee JW,Fang X,Dolganov G,et al.Acute lung injury oedema fluid decreases net fluid transport across human alveolar epithelial type Ⅱ cells[J].J Biol Chem,2007,282(33):24109-24119.
3 Eaton DC,Helms MN,Koval M.The contribution of epithelial sodium channels to alveolar function in health and disease[J].Annu Rev Physiol,2009,71:403-423.
4 Randrianarison N,Clerici C,Ferreira C,et al.Low expression of the beta-ENaC subunit impairs lung fluid clearance in the mouse[J].Am J Physiol Lung Cell Mol Physiol,2008,294(3):L409-416.
5 Speyer CL,Rancilio NJ,McClintock SD,et al.Regulatory effects of estrogen on acute lung inflammation in mice[J].Am J Physiol Cell Physiol,2005,288(4):C881-890.
6 Massaro D,Massaro GD.Estrogen regulates pulmonary alveolar formation,loss,and regeneration in mice[J].Am J Physiol Lung Cell Mol Physiol,2004,287(6):L1154-1159.
7 Laube M,Küppers E,Thome UH.Modulation of sodium transport in alveolar epithelial cells by estradiol and progesterone[J].Pediatr Res,2011,69(3):200-205.
8 Carey MA,Card JW,Voltz JW.The impact of sex and sex hormones on lung physiology and disease:lessons from animal studies[J].Am J Physiol Lung Cell Mol Physiol,2007,293(2):L272-278.
9 Deitch EA,Livingston DH,Lavery RF,et al.Hormonally active women tolerate shock-trauma better than do men:a prospective study of over 4000 trauma patients[J].Ann Surg,2007,246(3):447-453.
10 Ananthakrishnan P,Cohen DB,Xu DZ,et al.Sex hormones modulate distant organ injury in both a trauma/hemorrhagic shock model and a burn model [J].Surgery,2005,137(1):56-65.
11 Mendelsohn ME,Karas RH.Molecular and cellular basis of cardiovascular gender differences[J].Science,2005,308(5728):1583-1587.
12 Björnström L,Sjöberg M.Mechanisms of estrogen receptor signaling:convergence of genomic and nongenomic actions on target genes[J].Mol Endocrinol,2005,19(4):833-842.
13 Lang F,Shumilina E.Regulation of ion channels by the serum- and glucocorticoid-inducible kinase SGK1[J].FASEB J,2013,27(1):3-12.
14 Watt GB,Ismail NA,Caballero AG.Epithelial Na+channel activity in human airway epithelial cells:the role of serum and glucocorticoid-inducible kinase 1[J].Br J Pharmacol,2012,166(4):1272-1289.
15 Arroyo JP,Lagnaz D,Ronzaud C.Nedd4-2 modulates renal Na+-Cl-cotransporter via the aldosterone-SGK1-Nedd4-2 pathway[J].J Am Soc Nephrol,2011,22(9):1707-1719.
16 Williams DL,Ozment-Skelton T,Li C.Modulation of the phosphoinositide 3-kinase signaling pathway alters host response to sepsis,inflammation,and ischemia/reperfusion injury[J].Shock,2006,25:432-439.
17 Deng W,Li CY,Wang DX.Regulation of ENaC-mediated alveolar fluid clearance by insulin via PI3K/Akt pathway in LPS-induced acute lung injury[J].Respir Res,2012,13:29.
18 Soundararajan R,Pearce D,Ziera T.The role of the ENaC-regulatory complex in aldosterone-mediated sodium transport[J].Mol Cell Endocrinol,2012,350(2):242-247.
19 Mansley MK,Wilson SM.Effects of nominally selective inhibitors of the kinases PI3K,SGK1 and PKB on the insulin-dependent control of epithelial Na+absorption[J].Br J Pharmacol,2010,161(3):571-588.
猜你喜欢
中国药学药品知识仓库(2022年1期)2022-03-23
昆明医科大学学报(2022年1期)2022-02-28
湖南畜牧兽医(2021年6期)2022-01-24
食品安全导刊(2021年21期)2021-08-30
天津医科大学学报(2021年4期)2021-08-21
现代临床医学(2021年4期)2021-07-31
天津医科大学学报(2019年6期)2019-08-13
中国人兽共患病学报(2018年7期)2018-07-31
中国组织化学与细胞化学杂志(2017年1期)2017-06-15
中成药(2017年3期)2017-05-17
