
微波辅助合成甲氧羰基-β-环糊精碳酸酯
2014-02-05 02:38张毅民刘红莎张惠莹朱艳芳
化学工业与工程 2014年4期
张毅民,刘红莎,张惠莹,朱艳芳
(1.天津大学化工学院,绿色合成与转化教育部重点实验室,天津300072;2.天津大学管理与经济学部,天津300072)
β-环糊精(β-CD)是由淀粉经酶降解得到的一类由7个D-吡喃型葡萄糖单体经α-1,4糖苷键连接而成的中空环状结构的低聚糖。因其具有内疏水外亲水的独特结构以及空腔内径大小适中,包结能力强,经济易得等优点而得到广泛应用。然而,由于β-CD水溶性较差,极大地限制了它在药物制剂[1]和模拟酶[2]等多方面的应用。 为了克服这一缺点,国内外许多研究者对其进行了羟基衍生化,得到了一系列β-CD衍生物。其中,用甲基化试剂对β-CD上的羟基进行甲基化后所得的甲基-β-CD,其水溶性增大了几十倍,抗水解性和包结能力也较母体得到明显地提高[3];用环氧丙烷对 β-CD进行羟丙基化后,所得的2-羟丙基-β-CD在水中的溶解度超过 50 g,溶血性比母体更低[4]。 对 β-CD进行硫酸酯化后,所得β-环糊精硫酸酯具有极高的水溶性,在对映体的拆分中有较好的效果[5-6]。同时,还有广泛的生物活性,如抑制肿癌新生血管形成[7]。对β-CD碳酸酯化后,所得的β-环糊精碳酸酯,不仅可以固定在膜反应器上催化水解酯类化合物[8-9],而且可以作为手性选择体用于毛细管电泳分离对映体[10]。
文献 [11]报道了乙氧基羰基-γ-CD(γ-CDOCOOCH2CH3)的合成方法、结构表征以及用作药物(尤其是酸性药物)增溶剂等内容。文献[12]用DBU有机强碱作催化剂,在二甲基亚砜(DMSO)溶剂中,以碳酸二甲酯(DMC)与β-CD进行甲氧羰基化反应,制得β-环糊精碳酸酯。该产物是在0.1 MPa和60℃条件下反应6~8 h后,减压蒸出溶剂,再用丙酮沉淀出。这种方法虽然可以制备出β-环糊精碳酸酯,但所用的溶剂DMSO有毒、易分解,后处理复杂,而且反应时间长。因此,研究者开始寻找新型的制备方法。新兴的微波辐射技术因具有能耗低、反应时间短、产率高、选择性好等优点而在有机合成中得到应用与发展[13]。而目前关于β-CD碳酸酯化的微波反应研究却很少,所以笔者以碳酸二甲酯作羰基化试剂在 N,N-二甲基甲酰胺(DMF)溶剂中,采用微波辐射技术快速地制备了甲氧羰基-β-环糊精碳酸酯衍生物,并进一步分析了其反应机理。
1 实验部分
1.1 仪器与试剂
高效液相色谱仪(LabA llaince Pc2001二元梯度系统),seriesⅢ型泵,RI Detector,用十八烷基硅烷键合硅胶为填充剂的 Kromasil色谱柱。测试条件为:以水-甲醇(80∶20)为流动相;柱温 35℃,流速0.6 m L/m in,进样量2μL;MAS-Ⅱ型常压微波合成/萃取反应工作站(上海新仪微波化学科技有限公司);Perkin-Elmer型傅里叶红外光谱分析仪(美国Perkin-Elmer公司),波长范围 400~4 000 cm-1;LCQ Advantage MAX型液相色谱质谱仪(美国菲尼根公司),检测范围 m/z为0~2 000 cm-1;INOVA 500 MHZ超导核磁共振谱仪(美国Varian公司),磁场强度为11.75 TSLA,磁场漂移≤3 h。
β-CD和DMC购于天津市光复精细化工研究所;DMF购于科安隆博华(天津)医药化学有限公司;无水 K2CO3购于天津市博迪化工股份有限公司;丙酮购于天津市江天化工技术有限公司;甲醇、正丙醇、乙酸乙酯、氨水购于天津市康科德科技有限公司。其中甲醇为色谱纯试剂,DMC、DMF、丙酮、正丙醇、乙酸乙酯、氨水为分析纯试剂,所有试剂均未经处理,直接使用。
1.2 合成方法
称量15 gβ-CD置于300 m L长颈三口烧瓶中,再量取90 m L DMF加入其中,电磁搅拌至β-CD全部溶解,溶液澄清,再加入0.3 g无水碳酸钾固体小颗粒。将此混合溶液置于微波合成反应器中,设定反应温度90℃,微波功率400 W,转速600 r/m in,在冷凝条件下,边滴加31 m L DMC(10 min滴加完毕),边在微波辐射下反应21 m in。反应结束后,静置,待反应液冷却到室温,将上清液转移至单口烧瓶,减压旋蒸除去溶剂DMF和未反应的DMC,真空度控制在-0.1 MPa,温度范围为65~83℃。浓缩至浅黄色黏稠液时,加入50 m L丙酮,磁力搅拌12 h至产品均匀地分散在丙酮中。过滤除去丙酮,将湿产品置于真空干燥箱干燥12 h,最后得到浅黄色或白色固体颗粒约17.32 g。产品经 TLC测定(展开剂 m(正丙醇)∶m(乙酸乙酯)∶m(水)∶m(氨水)为6∶1∶3∶1)[14]有 4 个点,比移值分别为 0.22,0.36,0.46和0.50,第1个斑点为 β-CD,其余斑点为不同取代度的β-CD衍生物。
1.3 反应机理
根据详细分析推测,用脂类的O-羰基化方法合成甲氧羰基-β-环糊精碳酸酯的总反应方程式和反应机理如图1。
K2CO3做碱性催化剂作用于 β-CD的羟基,使羟基失去质子成为氧负离子。氧负离子很活泼,在低温下主要进攻 DMC上的羰基碳原子,形成过渡态,DMC失去甲氧基,β-CD得到甲氧基羰基形成甲氧羰基-β-环糊精碳酸酯,并重新生成 K2CO3。DMC的副反应产物为CH3OH和CO2。这一机理属于固液相转移催化剂(PTC)条件下的酯交换范畴。
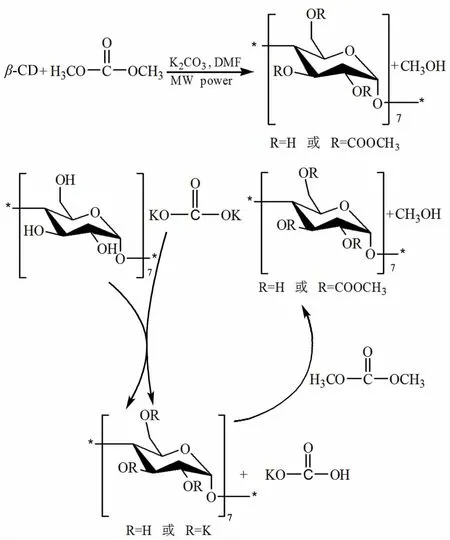
图1 甲氧羰基-β-环糊精碳酸酯总反应方程式和反应机理Fig.1 The total reaction equation and m echanism of m ethoxycarbonyl-β-cyclodextrin carbonate
在使用微波辐射技术时,虽然整个 β-CD分子很大,处于微波电场中不能转动或者转移,但 β-CD羟基可以局部扭转,然而,不能瞬时顺应微波电场的方向以改变排列取向,所以此交变过程就会产生摩擦,转化为热能深入到分子内部,产生了“热效应”,提高了 β-CD分子的温度,从而导致 O-H键断裂,加速生成 β-CD氧负离子[15-16]。 另外,与常规加热不同,微波有“特殊效应”可以降低PTC酯交换反应的吉布斯自由能;还可以在体系中形成“过热效应”,使溶剂温度比常规加热温度高,提高反应速率;K2CO3颗粒处于电场中时,被加热的速度和温度比周围介质更快、更高,会在表面形成“热点”而得到活化,并且其存在可以促进反应物吸热,使体系快速升温,且温度分布均匀[17-19]。因此,该反应可以在温和的条件下快速完成。
2 结果与讨论
β-CD在无溶剂DMF(β-CD悬浮在与溶剂 DMF等体积的DMC中)或者无催化剂条件下与DMC不反应,用薄层色谱法(TLC)检测均无甲基化产物生成。使用本研究的方法合成的产物经过高效液相色谱、质谱、红外、核磁共振等手段表征,证明生成了新型产物甲氧羰基-β-环糊精碳酸酯。
2.1 高效液相色谱法(HPLC)测含量
采用外标法做 β-CD标准曲线,以峰面积计算测定产物中β-环糊精的剩余量,得到原料的转化率为93.22%,即粗产品中原料β-CD与产物的质量比约为 7∶93。
2.2 电喷雾质谱(ESI-M S)
β-CD衍生物的取代度采用每个β-CD分子(共21个羟基)被取代基取代的个数来表示[20](因此最大值为21),通过质谱测定。甲氧羰基-β-环糊精碳酸酯质谱如图2。
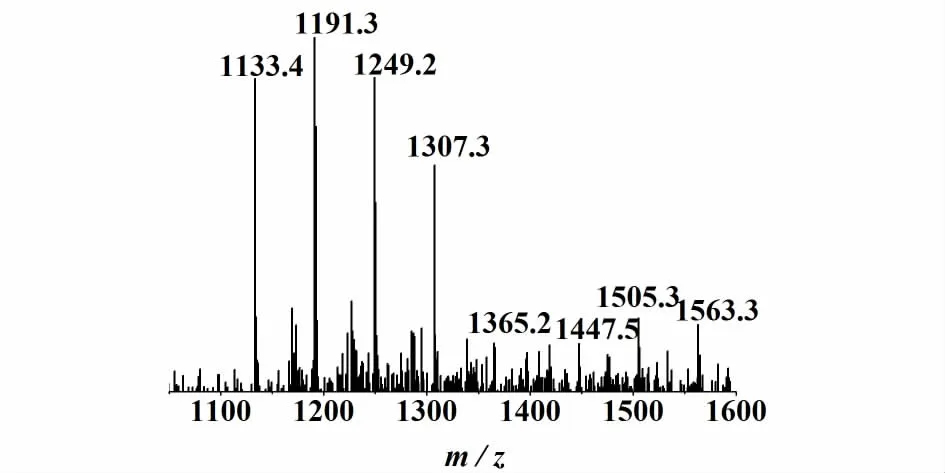
图2 甲氧羰基-β-环糊精碳酸酯的质谱图Fig.2 M S spectrum of m ethoxycarbonyl-β-cyclodextrin carbonate
质谱分析采用负离子模式,谱图中出现8个主要信号,主要碎片峰为m/z=1191.3。1 133.4是β-环糊精的分子离子峰,1个甲氧基羰基的相对分子质量是59,每被1个甲氧基羰基取代,β-环糊精碳酸酯的相对分子质量增加59-1=58,若包含结晶水需要再加上18的倍数。由1133+58=1191,所以可以判断1 191.3处的峰是取代度为1的甲氧羰基-β-环糊精碳酸酯的准分子离子峰;同理,1 249.2处是取代度为2的甲氧羰基-β-环糊精碳酸酯的准分子离子峰;1 307.3、1 365.2、1 505.3、1 563.3 处分别是取代度为 3、4、5、6 的甲氧羰基-β-环糊精碳酸酯的准分子离子峰。而甲酸相对分子质量为46,1365+46+2 ×18+58=1505,故 1 505.3、1 563.3处分子离子峰为取代度为5、6的甲氧羰基-β-环糊精碳酸酯。由甲氧羰基-β-环糊精碳酸酯的取代度和其对应丰度可求出平均取代度为2.423。MS(%):1 191.3(100)、1 249.2(88.71)、1 307.2(64)、1 365.2(13.69)、1 505.3(20.73)、1 563.3(18.97)。
2.3 红外吸收光谱(IR)
用KBr将β-CD和甲氧羰基-β-环糊精碳酸酯分别压片,测定红外光谱图如图3。
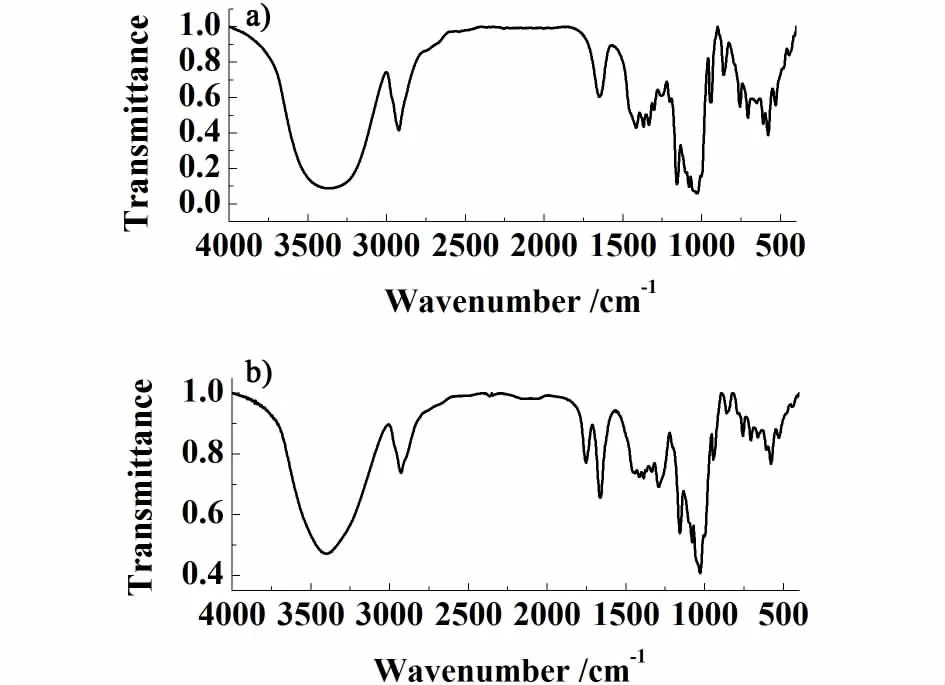
图3 β-环糊精a)及其碳酸酯b)的红外吸收光谱图Fig.3 IR spectrum ofβ-cyclodextrin a)and m ethoxycarbonyl-β-cyclodextrin carbonate b)
β-CD的主要红外吸收峰波数(cm-1)为3 367,2 924,1 649,1 416,1 253,1 157,1 080,1 030,939,578,甲氧羰基-β-环糊精碳酸酯主要红外吸收峰波数(cm-1)为 3 400,2 928,1 753,1 664,1 460,1 387,1 292,1 157,1 080,1030,945,578。 在指纹区,1 157 cm-1和939 cm-1(945 cm-1)特征峰归属于C-O键伸缩振动,1 080 cm-1和1 030 cm-1是 β-CD吡喃环上 C-O单键伸缩振动峰,1 649 cm-1(1 664 cm-1)为分子内键合水特征峰[21],3 367 cm-1(3 400 cm-1)归属于羟基特征峰。对比β-CD和 β-CD衍生物的红外谱图可以发现:吡喃糖环特征峰(1 500~400 cm-1)只有微小变化,证明 β-CD衍生物与β-CD具有相同的骨架。β-CD衍生物红外谱图中只是很明显地新增加了1 753 cm-1典型羰基峰和1 460 cm-1甲基C-H弯曲振动峰,证明β-CD衍生物为 甲 氧 羰 基-β-环 糊 精 碳 酸 酯, 结 构 为 β-CDOCOOCH3,而且1 753 cm-1典型羰基峰证明得到的甲氧羰基-β-环糊精碳酸酯呈直链状[22]。
2.4 核磁共振谱(NM R)
以 DMSO-d6为溶剂,产物的1H-NMR及13CNMR见图4和图5。
从产物的1H-NMR谱图分析得知:δ5.50~6.00为 C-2位与C-3位未取代H-O,δ4.25~4.75为C-6位未取代 H-O。 δ4.82(s,7H)为 C-1位 H,其反应前后数目不变。因此根据平均取代度计算公式:

得到产物平均取代度为2.567。此值与质谱测定的平均取代度基本一致。

图4 β-环糊精 a)及其碳酸酯b)的1 H-NM R谱图Fig.4 1 H-NM R spectrum ofβ-cyclodextrin a)and m ethoxycarbonyl-β-cyclodextrin carbonate b)

图5 β-环糊精 a)及其碳酸酯b)的13 C-NM R谱图Fig.5 13 C-NM R spectrum ofβ-cyclodextrin a)and m ethoxycarbonyl-β-cyclodextrin carbonate b)
查阅13C-NMR化学位移表可以确认图5中δ 155.44为碳酸酯羰基碳化学位移,与文献报道一致[23],δ55.15 为 CH3O-化学位移,产物 δ102.41(C-1),82.03(C-4),73.53(C-3),72.89(C-2),72.51(C-5),60.41(C-6)与未反应的 β-CD相应碳的化学位移相比在 δ102.62(C-1),82.20(C-4),73.73(C-3),73.08(C-2),72.71(C-5),60.59(C-6)处都稍微偏向低场,推测应该是取代基的去屏蔽效应。β-CD中C-6被取代后的化学位移会向低场移动约 Δδ5~10,通常在 δ66.4 ~71.3 范围内[24-25],而在图4中δ70.00附近的化学位移即为取代后的C'-6,说明取代主要发生在 C-6位,原因可能是C-6位的伯羟基位阻小,容易被大基团取代。
3 结论
在微波辐射工艺和催化剂辅助下,用绿色试剂DMC和β-CD成功地合成了甲氧羰基-β-环糊精碳酸酯。此新工艺过程简单、快速、高效,使反应时间由传统方法的十几小时减少至数分钟,大大缩短了反应周期,提高了效率;而且所用试剂DMC无毒,符合绿色化学的发展方向。通过高效液相色谱法测定了产物中β-CD的相对含量,得到原料转化率为93.22%,质谱法测定产物平均取代度为2.423,采用红外吸收光谱和核磁共振波谱证明了其化学结构,鉴定产物中含有C=O和-OCH3基团,结构为β-CD-OCOOCH3。
致谢:
感谢陈立功老师课题组为本实验提供微波反应仪器,同时感谢分析中心乔老师与周老师给予质谱和核磁谱知识指导。
[1]Szente L,Szejtli J.Highly soluble cyclodextrin derivatives:Chemistry,properties,and trends in development[J].Advanced Drug Delivery Reviews,1999,36(1):17-28
[2]Tabushi I.Cyclodextrin catalysis as amodel for enzyme action [J].Accounts of Chem ical Research,1982,15(3):66-72
[3]M ino R C,Vivienne J G,Luigi R N,et al.Unusual
1C4conformation of a methylglucose residue in crystalline permethyl-β-cyclodextrin monohydrate[J].Journal of the Chemical Society,Perkin Transactions 2,1994,(10):2 071-2 072
[4]袁超,金征宇.响应面法优化羟丙基-β-环糊精制备工艺[J].食品科学,2007,28(3):147-151 Yuan Chao,Jin Zhengyu.Optimization of hydroxypropyl-β-cyclodextrin preparation process by response surface method[J].Food Technology,2007,28(3):147-151(in Chinese)
[5]杨艳丽.β-环糊精衍生物的合成及其在毛细管电泳手性拆分中的应用[D].山东:山东大学,2009 Yang Yanli.Synthesis ofβ-cyclodextrin derivatives and application for enantioseparation in capillary electrophoresis[D].Shandong:Shandong University,2009(in Chinese)
[6]孙青,刘长海,李国栋,等.β-环糊精硫酸酯的合成及其在舒必利毛细管电泳拆分中的应用[J].第二军医大学学报,2004,25(11):1 259-1 260 Sun Qing,Liu Changhai,Li Guodong,et al.Synthesis ofβ-cyclodextrin sulfate and its application in enantiomeric separation of sulpiride by capillary zone electrophoresis[J].Academ ic Journal of Second M ilitary Medical University,2004,25(11):1 259-1 260(in Chinese)
[7]仇毓东,陈汉,林川,等.β-CD14S抑制大鼠肝癌新生血管形成的实验研究[J].南京大学学报:自然科学,2002,38(5):681-685 Qiu Yudong,Chen Han,Lin Chuan,et al.Inhibition of angiogenesis on HCC in rat byβ-CD-14S [J].Journal of Nanjing University:Natural Science,2002,38(5):681-685(in Chinese)
[8]Natoli M,Pagliero C,Trotta F,et al.A study of catalyticβ-cyclodextrin carbonatemembrane reactor performance in PNPA hydrolysis[J].Journal of Molecular Catalysis A:Chemical,1997,121(2/3):179-186
[9]Gordano A,Trotta F,Manferti C,et al.Catalytic behaviour of carbonateβ-CD entrapped in PEEK-WC membranes[J].Journal of Inclusion Phenomena and Macrocyclic Chemistry,2002,44(1/4):433-437
[10]Zerbinatia O,Trotta F,Giovannolia C,et al.New derivatives of cyclodextrins as chiral selectors for the capillary electrophoretic separation of dichlorprop enantiomers[J].Journal of Chromatography A,1998,810(1/2):193-200
[11]Fenyvesi E,Szejtli J,Trotta F,et al.Comparison of the solubilizing effect of ethyl carbonate ofγ-cyclodextrin to other cyclodextrin derivatives,Santiago de Compostela,Spain,May31-June3,1998[C]//Proceedings of the Ninth International Symposium on Cyclodextrins.The Netherlands:Kluwer Academic Publishers,1999:289-292
[12]肖璐.β-环糊精酯类衍生物的合成[D].浙江:浙江大学,2005 Xiao Lu.The synthesis ofβ-cyclodextrin ester derivatives[D].Zhejiang:Zhejiang University,2005(in Chinese)
[13]Brett A R,Christopher R S.Toward rapid,“green”,predictable microwave-assisted synthesis[J].Accounts of Chemical Research,2005,38(8):653-661
[14]Jindrich J,Pitha J,Lindberg B,et al.Regioselectivity of alkylation of cyclomaltoheptaose(β-cyclodextrin)and synthesis of its mono-2-O-methyl,-ethyl,-allyl,andpropyl derivatives[J].Carbohydrate Research,1995,266(1):75-80
[15]Chen S,Wu C,Wu S,et al.The studies ofm icrowave effects on the chemical reactions[J].Journal of the Chinese Chem ical Society,1997,44(3):169-182
[16]Singh V,Tiwari A.M icrowave-Accelerated methylation of starch[J].Carbohydrate Research,2008,343(1):151-154
[17]Perreux L,Loupy A.A tentative rationalization of microwave effects in organic synthesis according to the reaction medium,and mechanistic considerations[J].Tetrahedron,2001,57(45):9 199-9 223
[18]Galema S A.M icrowave chemistry[J].Chem ical Society Reviews,1997,26(3):233-238
[19]Lidström P,Tierney J,Wathey B,et al.Microwave assisted organic synthesis-A review[J].Tetrahedron,2001,57(45):9 225-9 283
[20]Challa R,Ahuja A,Ali J,et al.Cyclodextrins in drug delivery:An updated review[J].AAPS Pharm Sci Tech,2005,6(2):329-357
[21]Fanga J M,Fowlera P A,Tomkinsona J,et al.The preparation and characterisation of a series of chemically modified potato starches[J].Carbohydrate Polymers,2002,47(3):245-252
[22]Kennedy J F,Tun H C.Preparation of carbonates of polysaccharides and cycloamyloses[J].Carbohydrate Research,1973,26(2):401-408
[23]Trotta F,Moraglio G,Zerbinati O et al.A lkyl capped carbonates ofβ-cyclodextrin [J].Journal of Inclusion Phenomena and Molecular Recognition in Chem istry,1996,23(4):269-276
[24]Sun H,Hao A,Yang Y,et al.New cyclodextrin derivative 6-O-(2-hydroxyl-3-betainylpropyl)-β-cyclodextrin:preparation and its app lication for enantiomer separation of drugs by capillary electrophoresis[J].Journal of Inclusion Phenomena and Macrocyclic Chemistry,2009,65(3/4):427-430
[25]Forgo P,D’Souza V T.Unambiguous identification of regioisomers in selectively modified β-cyclodextrins[J].Journal of Organic Chemistry,1999,64(1):306-309
猜你喜欢
陶瓷学报(2021年5期)2021-11-22
中成药(2018年8期)2018-08-29
中成药(2018年6期)2018-07-11
中成药(2018年4期)2018-04-26
湖北农业科学(2017年24期)2018-01-27
上海农业科技(2016年6期)2016-12-23
山东工业技术(2016年15期)2016-12-01
环境科技(2016年4期)2016-11-08
化工进展(2015年2期)2015-08-19
中国兽药杂志(2014年7期)2014-11-23
