
直接液流燃料电池的制备、活化及性能
2014-02-03 02:04韩大量黄成德
化学工业与工程 2014年3期
米 南,韩大量,黄成德*,张 晶
(1.天津大学化工学院,天津300072;2.中国电子科技集团第十八研究所,天津300381)
经过20年左右的研究与发展,直接甲醇燃料电池(DMFC)以其燃料来源丰富、结构简单、安全性能高、持续供电时间长等优点已经在民用便携产品以及军事移动性仪器等领域得到了一定的应用[1-3],但是DMFC中存在的一些重大问题如甲醇渗透、阳极氧化反应活性不高以及阴极水淹等问题也亟待从根本上加以解决[4-6]。近年来,Ilicic等提出了一种直接液流氧化还原燃料电池(DLRFC)并开展了许多相关研究[7-9],用以解决 DMFC电池中所存在的一系列问题。DLRFC以有机燃料如甲醇、甲酸等做阳极燃料,以氧化还原电对如Fe2+/Fe3+等做阴极氧化剂,通过电对还原与燃料氧化得失电子的过程向外界提供电能。在该构筑方式中,阴极和阳极均为液体进料,因此不存在阴极水淹的问题,并且两种液体混合时不会发生任何反应,因此避免了燃料渗透的影响。此外,由于阴极不需要再使用贵金属催化剂,因此大大降低了电池整体的成本,为其商业化发展提供了进一步的可能性。
但是Ilicic等构筑的直接液流燃料电池单体是采用碳纸作为阴极材料,通过对内置氮囊充气以保证阳极催化层与质子交换膜的紧密接触。这样的结构设计在一定程度上不仅增加了复杂性,也可能会因为阳极催化层与质子交换膜间结合不紧密而影响电子、离子的传输等。此外,由于使用多层碳纸作为电池阴极材料,较大的界面电阻也阻碍了电池性能的提高。在实验中,他们也未曾讨论过活化条件等对电池性能的影响。本研究采用热压法将阳极催化层与质子交换膜压制成阳极半膜电极(AHME)用于DLRFC中。在电池单元中去除了氮囊部件,简化了电池的整体构造,且阴极采用石墨毡材料。通过极化曲线测试分析了热压过程中的热压温度、热压压力以及热压时间对AHME性能的影响。此外,本研究还采用高温水、恒电流放电和变电流放电3种方式对DLRFC单体进行活化研究。并针对恒电流放电活化方式,进一步研究了恒电流活化时间、活化温度以及活化电流密度对电池性能的影响。
1 试验部分
1.1 主要试剂
Nafion膜溶液(DE520,质量分数为5%),上海河森电气有限公司;聚四氟乙烯乳液(PTFE,质量分数为 60%),Sigma-Aldrich公司;碳纸(TGP-H-060),Toray公司;PtRu/C催化剂[质量分数为30%, n(Pt)∶n(Ru)=2∶1],Johnson Matthey公司;碳粉(Vulcan XC-72),Carbot公司;Nafion 117 膜,上海河森电气有限公司;石墨毡(PAN基),北京市三业碳素有限公司;硫酸(质量分数为98%),天津科威公司;以下试剂均由天津光复精细化工研究所提供:甲醇,分析纯;硫酸亚铁,分析纯;硫酸高铁铵,分析纯;双氧水,分析纯;去离子水。
1.2 阳极半膜电极制备
1.2.1 阳极极片制备
将2×2 cm2碳纸在聚四氟乙烯乳液中浸泡5 min,然后在马弗炉中350℃煅烧1 h,自然冷却后取出备用。将碳粉与聚四氟乙烯乳液分散于异丙醇水溶液中超声分散1 h形成均匀的浆料,其中PTFE占固体总质量的20%。然后利用刮涂法将浆料均匀涂覆在已经处理好的碳纸层上,在80℃的真空干燥箱中烘干2 h,备用。将 PtRu/C催化剂与Nafion溶液分散于异丙醇与水组成的混合溶液中,超声分散1 h形成均匀的催化剂浆料,然后在真空干燥箱中干燥2 h,其中干态 Nafion占固体总质量的30%,PtRu金属载量为2 mg/cm2。通过刮涂的方式将催化剂浆料分多次平整涂覆在阳极扩散层的表面,在80℃的真空干燥箱中烘干,放入密封袋中,待用。
1.2.2 Nafion膜处理
将Nafion 117膜先用去离子水以80℃处理1 h,再用质量分数为5%的 H2O2和去离子水分别以80℃处理1 h,除去膜表面的有机物杂质。处理后的Nafion 117膜用1 mol/L的 H2SO4,80℃活化1 h,再用去离子水80℃处理1 h,除去多余H2SO4。
1.2.3 热压阳极半膜电极
开启压膜机系统,按照阳极在下面,处理好的Nafion 117膜在上面的顺序摆放好,即为待压制的AHME。用2块平滑钢板将待压制的AHME夹紧,并放到压膜机的中心位置,按照表1中的数值设置热压参数,依次制备 6块 AHME。将压制好的AHME自然冷却至室温,即得阳极半膜电极。

表1 不同AHM E电极的热压过程条件参数Table 1 Parameters in hot-pressing process of AHM E
1.3 电池组装
图1为直接液流氧化还原燃料电池结构示意图。
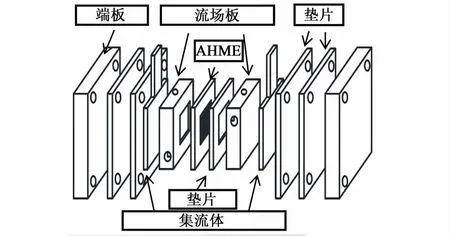
图1 直接液流氧化还原燃料电池结构图Fig.1 A schematic structure of direct liquid redox fuel cell
1.4 DLRFC的活化与性能测试
按照AHME1的热压条件制备6块性能相同的AHME,阴极采用10 mm厚的石墨毡,按照图1所示将各组件固定并夹紧。
分别采用如下3种方式对DLRFC电池单体进行活化。1)高温水活化:在阳极和阴极储液罐中分别装入500 mL蒸馏水,将储液罐放置于恒温水浴箱中加热,待蒸馏水温度稳定在80℃时,以2 m L/min的流速分别输送到电池阳极和阴极端,电池温度稳定在80℃,按此方式活化3 h。2)恒电流放电活化:阳极以2 m L/min的流速通入浓度为2 mol/L的甲醇溶液,阴极以5 m L/min的流速通入0.81 mol/L的 NH4Fe(SO4)2、0.09 mol/L的 FeSO4和0.5 mol/L的H2SO4的阴极电解液,保持电池温度为60℃,设置放电电流为20 mA/cm2,放电时间为3 h。3)变电流放电活化:阳极以2 m L/min的流速通入浓度为2 mol/L的甲醇溶液,阴极以5 m L/min的流速通入0.81 mol/L的 NH4Fe(SO4)2、0.09 mol/L的 FeSO4和0.5 mol/L的H2SO4的混合溶液,保持电池温度为60℃,设置放电电流分别为10、20和40 mA/cm2,每个阶段放电时间为1 h。
AHME和DLRFC电池单体的极化曲线采用如图2所示的自组装电池测试系统进行测试。阳极燃料液为甲醇溶液,阴极液为NH4Fe(SO4)2、FeSO4和H2SO4的混合溶液,电解液经过 BT01-DG2型蠕动泵(天津市协达伟业电子有限公司)循环更新,阳极燃料液的流速为2 m L/min,阴极电解液的流速为5 m L/min。电池的放电性能采用IT-8510型直流可编程电子负载(南京艾德克斯电子有限公司)与计算机联用进行测试,电池的负载电压不间断地从0.6 V变化到0.1 V,步长为0.02 V,每个工步稳定时间为1 min,由计算机自动记录每一个电压所对应的电流值。
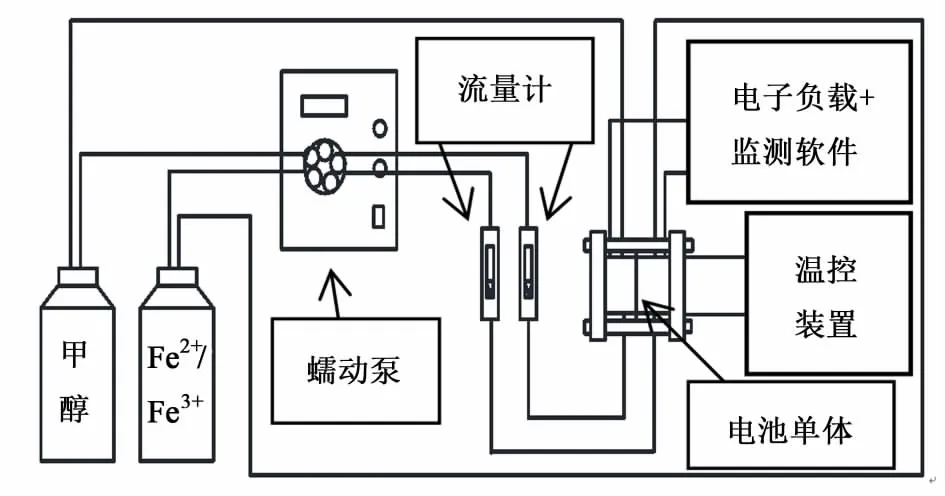
图2 DLRFC电池测试系统示意图Fig.2 Schematic of the DLRFC test system
2 结果与讨论
2.1 热压工艺的影响
为了研究热压过程中不同条件对阳极半膜电极电化学性能的影响,采用恒电流放电活化方式活化3 h后测试了AHME1~6的稳态电流-电压极化曲线,结果如图3a)~图3c)所示。
由图3a)可以看出,热压温度对AHME开路电压和电流密度的影响均不大,120和140℃热压制得的AHME开路电压均为0.46 V,电流密度均在100~105 mA/cm2之间。但是 120℃热压下的AHME在大电流密度下(>90 mA/cm2)性能略优于140℃热压的效果。产生这种现象的原因可能是当温度超过Nafion 117膜的玻璃化转变温度时,膜中所含有的团簇将解体并转化为玻璃态,温度越高,团簇解体越多,玻璃化程度越严重[10],膜的电导率会降低[11]。图3b)反映了热压压力对 AHME性能的影响。适当增大热压压力加大了催化层Nafion聚合物团粒的变形,使Nafion与催化层的接触界面增多,因而催化剂层活性比表面积增加。而且,压力增大还可以改善Nafion膜与催化层之间、催化剂层与扩散层之间的接触,减小层与层间的界面接触电阻[12],从而改善了AHME的放电性能。图3c)所示的是热压时间对AHME性能的影响,若热压时间太短,则 AHME的性能不稳定。当热压时间超过120 s以后,再延长时间对电池性能的影响并不大。
为了进一步分析热压时间的影响,对不同热压时间制得的 AHME进行了电化学阻抗谱测试,图3d)为相应的 Nyquist图。由图3d)可以看出,120 s和180 s热压AHME的阻抗谱在低频区差别较小,而60 s热压的AHME与二者差别较大。并且,120 s热压AHME的欧姆电阻为3.356Ω,而热压60 s和180 s后AHME的电阻分别为4.487和4.677Ω,这可能是由于热压时间太短,催化剂层中的Nafion聚合物来不及达到软化状态[13],以致于Nafion和催化层的结合不好,导致AHME内部电阻变大。而进一步延长热压时间会增加电极压缩率,使得阳极半膜电极内催化层和扩散层更加致密,一定程度上阻碍了电极内的物质传递[13]。
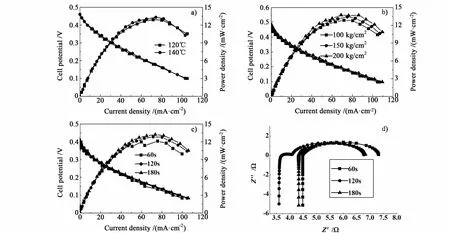
图3 热压条件对AHM E性能的影响.a)热压温度,b)热压压力,c)热压时间,d)不同热压时间AHM E的交流阻抗谱Fig.3 Effect of hot-pressing conditions on AHM E.a)Hot-pressing tem perature,b)Hot-pressing pressure,c)Hot-pressing time and d)EIS of AHM E hot pressed for different time
2.2 同活化方式的比较
由于通过热压法制备的 AHME,可能会将催化层中某些催化剂封入死区,因此对于一个新组装的DLRFC单体,必须通过一定的活化过程才能使电池单体达到最佳性能[14-16]。 因此,本研究采用高温水、恒电流放电和变电流放电3种方式对DLRFC单体进行活化研究。
比较以3种活化方式活化3 h后DLRFC的开路电压,由图4发现恒电流活化后电池的电压为0.46 V,低于高温水活化后的0.50 V和变电流活化的0.48 V。这可能是由于电流恒定,电极内活性点较少,因此相应的电压也较低。但是放电活化后电池能达到较高的电流密度,这主要是因为在欧姆极化控制区域,放电活化方式下的放电曲线比高温水活化下的放电曲线斜率更小。分析可能的原因是在阳极区,恒电流活化和变电流活化在活化过程更有利于干膜吸水以及促进膜内物质通道的形成[15],而高温水活化只能促进干膜吸水,却未能打通质子传递通道。并且在阴极区,随着活化的进行阴极电解液逐渐进入石墨毡内部,增强了与电极的接触。从图4中还可以看出,变电流活化方式能使DLRFC达到的最大电流密度为109.5 mA/cm2要好于恒电流活化后的104.7 mA/cm2,这可能是在相同时间内变电流活化可以使阳极区中更多的催化剂得到活化,每次改变电流都提高了膜内的水含量。
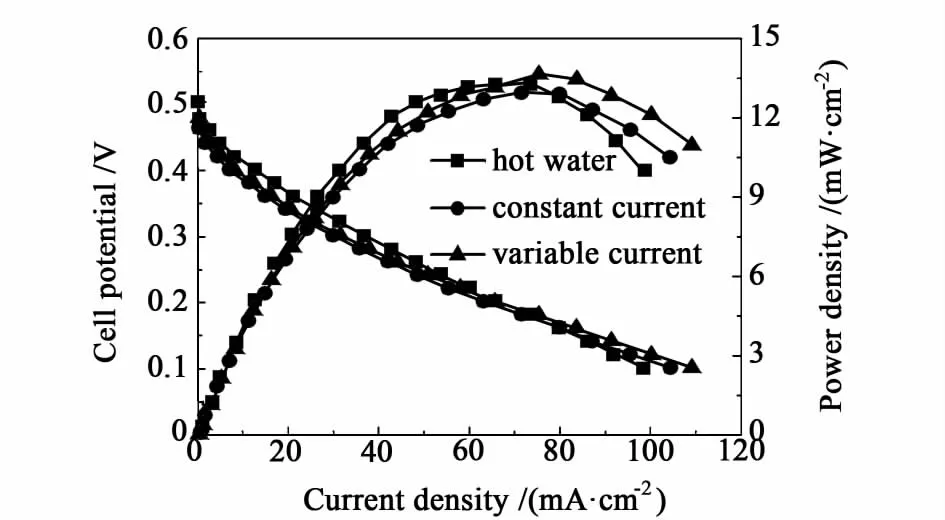
图4 不同活化方式的比较Fig.4 Com parison of the three activation methods
2.3 恒电流活化工艺的比较
图5a)反映了DLRFC单体通过不同时间的恒电流活化后能达到的效果。
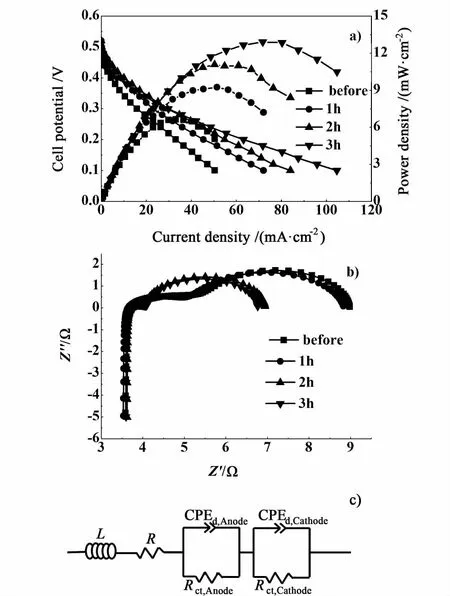
图5 不同活化时间对电池性能的影响a)电流-电压曲线,b)Nyquist图,c)等效电路图Fig.5 Performance of DLRFC activated by variab le time a)I-V curves, b)Nyquist p lot, c)EIS equivalent circuit
由图5可以看出,活化时间越长,活化后电池性能越好。这可能是因为随着活化的进行,Nafion膜中的全氟磺酸基团被润湿,全氟磺酸基团在吸水后膨胀与更多的Pt接触形成反应活性点[17-18],参与电化学反应,从而使性能提高。比较未活化和活化3 h后DLRFC单体的极化曲线,发现前者由浓差扩散引起的极化非常严重,这说明阳极区内催化层内的水分不足。而活化一定时间后,这种浓差极化已经得到了很大的缓解。
为了进一步分析活化时间对电池性能产生影响,对活化时间不同的电池进行电化学交流阻抗测试,测试结果如图5b)所示,可见电池的Nyquist曲线均呈拖着1条尾巴的2个连续的下沉半圆弧形。
根据 Muller等[19]的研究,由图5b)可以看到,活化时间不同的DLRFC单体高频阻抗弧比较相近,说明电池的欧姆电阻并没有太大的区别;随着活化时间延长,中频弧和低频弧均呈减小的趋势,这说明DLRFC中的电化学极化电阻和阳极催化层内的传质电阻变小。活化2 h和活化3 h电池的交流阻抗谱很相近,表明对于DLRFC,恒电流活化2~3 h即可使其达到稳定的状态。
采用ZSimpW in软件对图5b)的交流阻抗图进行拟合,得到如图5c)所示的等效电路图。图5中,R代表整个电池的欧姆电阻,Rct,Anode和 CPEd,Anode是用于模拟阳极的元件,分别代表阳极甲醇电氧化过程中的电荷传递电阻和电容特性;Rct,Cathode和 CPEd,Cathode是用于模拟阴极的元件,分别代表阴极Fe2+/Fe3+电对还原过程中的电荷传递电阻和电容特性。拟合后,等效电路中各元件对应的数值如表2所示。
从表2可见,电池的电感 L基本一致,均在8.2×10_6H左右,这说明电池导线元件的连接基本相同。比较活化不同时间电池的Rct值发现,活化时间越长,Rct,Anode和 Rct,Cathode值均越小。 Rct代表在氧化还原反应中,电化学反应过程中电荷传递阻力。在电池内部,影响 Rct,Anode大小的因素来源于Nafion膜传导质子能力的大小以及催化层内电子得失的快慢,影响 Rct,Cathode的因素为阴极区电解质与石墨毡内部接触面积的大小。表2的结果说明适当延长活化时间既有利于打通质子交换膜中的传递通道,也促进了电解质与石墨毡的进一步接触。
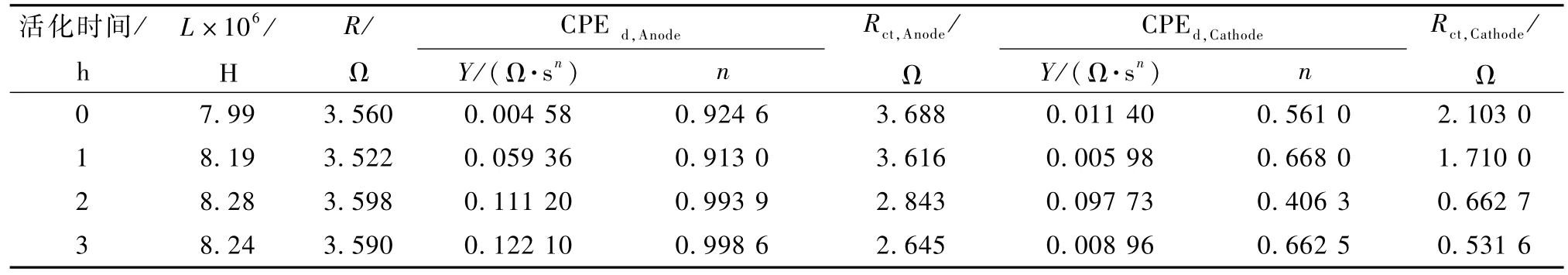
表2 不同活化时间下M EA的交流阻抗拟合值Table 2 The fitted values of EIS during the d ifferent activation time
从图6a)中可以看出,在60℃下恒电流活化3 h的电池性能最优,最大功率密度能达到13.5 mW/cm2,高于40和80℃下恒电流活化后的电池性能。这可能是因为活化温度的升高有利于向阳极区引入更多的水蒸气,并且加剧水分子的热运动,促使催化层的Nafion进一步润湿以及水分子在催化层和Nafion膜内的均匀分布,并且提高了阴极区石墨毡与电解质的反应速度,这些均可能促进电池反应电流的增大。但是温度过高,容易引入过量的水蒸气使得催化层内的 Nafion膨胀速度加快,一旦超过了Nafion膜的膨胀速度,将导致催化层与膜之间部分质子通道断裂,使电池性能反而下降。因此,DLRFC单体活化温度为60℃最适宜。而采用不同电流密度对电池进行连续恒电流放电活化的结果图6b)发现,当电流密度增大到一定程度之后,活化使电池性能改善的效果有所下降。由于阳极使用的是Pt-Ru/C催化剂,并且当阳极电位高于0.6 V时,Pt-Ru/C催化剂的 Ru逐渐溶解转变成 Pt/C使电极反应的电位升高[20]。因此,采用大电流密度放电时,阳极过电位很可能会随着放电电流密度的增大而提高,致使 Ru发生溶解,从而导致 DLRFC性能下降。
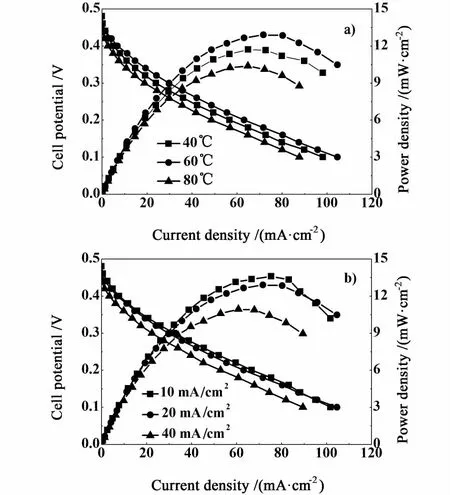
图6 恒电流活化工艺对电池性能的影响a)恒电流活化温度,b)恒电流活化电流密度Fig.6 Effect of constant cur rent activation cond itions on DLRFC a)Tem perature,b)Current density
3 结论
1)通过比较不同热压条件制备的AHME的极化曲线,我们发现最适宜热压温度为120℃,热压压力为200 kg/cm2,热压时间为120 s。
2)比较高温水、恒电流放电与变电流放电活化3种活化工艺活化DLRFC后的极化曲线发现,3种方式均可以降低电池内部的电阻,但是高温水活化效果没有放电活化效果好。
3)通过对恒电流活化方式中不同活化时间DLRFC的极化曲线及EIS测试发现,适当延长活化时间有利于打通质子交换膜中的传递通道以及阴极电解液与石墨毡的进一步接触。适当提高活化温度,有利于促使催化层的Nafion进一步润湿以及水分子在催化层和Nafion膜内的均匀分布。采用大电流密度放电时,很可能会使阳极过电位随着放电电流密度的增大而提高,造成Ru发生溶解,致使电池放电时阳极活化过电位增大,从而导致DLRFC性能下降。
[1]Chen C, Cha H.Strategy to optimize cathode operating conditions to improve the durability of a directmethanol fuel cell[J].Journal of Power Sources, 2012, 200:21_28[2]Reshetenko T V,Kim H T,Krewer U,et al.The effect of the anode loading and method of MEA fabrication on DMFC performance[J].Fuel Cells, 2007, 7(3):238_245
[3]Xue X,Bock C,Birry L,et al.The influence of Pt loading,support and Nafion content on the performance of directmethanol fuel cells:Examined on the example of the cathode[J].Fuel Cells, 2011, 11(2):286_300
[4]Li X,Faghri A.Development of a directmethanol fuel cell stack fed with puremethanol[J].International Journal of Hydrogen Energy, 2012, 37:14 549_14 556
[5]Cha H,Chen C,Wang R,et al.Performance test and degradation analysis of direct methanol fuel cell membrane electrode assembly during freeze/thaw cycles[J].Journal of Power Sources, 2011, 196:2 650_2 660
[6]Bae S,Kim S,Um S,et al.A prediction model of degradation rate formembrane electrode assemblies in direct methanol fuel cells[J].International Journal of Hydrogen Energy, 2009, 34:5 749_5 758
[7]Ilicic A,Dara M,Wilkinson D,et al.Improved performance of the directmethanol redox fuel cell[J].Journal of Applied Electrochemistry, 2010, 40:2 125_2 133
[8]Ilicic A,W ilkinson D,Fatih K,et al.High fuel concentration direct-liquid fuel cell with a redox coup le cathode[J].Journal of The Electrochemical Society,2008,155(12):B1 322_B1 327
[9]Ilicic A,Wilkinson D,Fatih K.Advancing direct liquid redox fuel cells:M ixed-Reactant and in situ regeneration opportunities[J].Journal of the Electrochemical Society, 2010, 157(4):B529_B535
[10]Zawodzinski T, Derouin C, Radzinski S, et al.Water uptake by and transport through Nafion®117 membranes[J].Journal of the Electrochemical Society, 1993, 140(4):1 041_1 047
[11]Lin J, Lai C, Ting F, et al.Influence of hot-pressing temperature on the performance of PEMFC and catalytic activity[J].Journal of Applied Electrochemistry, 2009,39:1 067_1 073
[12]Ting F, Lin J, Lai C, et al.Electrochemical impedance spectroscopy to evaluate the effect of pressure exerted in the hot-pressing stage on the performance of PEMFC[J].International Journal of Electrochemical Science,2012,7:7 165_7 178
[13]Yazdanpour M,Esmaeilifar A,Rowshanzami S.Effects of hot pressing conditions on the performance of Nafion membranes coated by ink-jet printing of Pt/MW CNTs electrocatalyst for PEMFCs[J].International Journal of Hydrogen Energy, 2012, 37:11 290_11 298
[14]Silva V, Rouboa A.Hydrogen-Fed PEMFC:Overvoltage analysis during an activation procedure[J].Journal of Electroanalytical Chemistry, 2012, 671:58_66
[15]Zhao X,LiW,Manthiram A.Comparison of the membrane-electrode assembly conditioning procedures for directmethanol fuel cells[J].Journal of Power Sources,2012,201:37_42
[16]Silva V, Rouboa A.In situ activation procedures applied to a DMFC:Analysis and optimization study[J].Fuel, 2012, 93:677_683
[17]Yu J, Matuura T, Yoshikawa Y, et al.In situ analysis of performance degradation of a PEMFC under nonsaturated humidification[J].Electrochemical and Solid State Letters, 2005, 8(3):A156_A158
[18]Yu J, Matsuura T, Yoshikawa Y, et al.Lifetime behavior of a PEM fuel cell with low humidification of feed stream[J].Physical Chemistry Chemical Physics,2005,7(2):373_378
[19]Mueller J, Urban P.Characterization of directmethanol fuel cells by AC impedance spectroscopy[J].Journal of Power Sources, 1998, 75(1):139_144
[20]Urban P, Funke A, Mueller J, et al.Catalytic processes in solid polymer electrolyte fuel cell systems[J].Applied Catalysis A:General, 2001, 221:459_470
猜你喜欢
中国特种设备安全(2022年6期)2022-09-20
制造技术与机床(2019年8期)2019-09-03
模具制造(2019年3期)2019-06-06
电子制作(2018年12期)2018-08-01
制造业自动化(2017年2期)2017-03-20
电镀与环保(2016年3期)2017-01-20
电镀与环保(2016年3期)2017-01-20
电镀与环保(2016年2期)2017-01-20
电源技术(2015年9期)2015-06-05
