
儿童Alport 综合征COL4A5 基因4 种新突变分析
2013-12-23 04:22黄建萍赵晓艳肖丽丽张尼娜
中国循证儿科杂志 2013年1期
都 娟 黄建萍 赵晓艳 王 硕 肖丽丽 张尼娜
Alport 综合征(AS)是儿童期以血尿为首发症状起病的常见疾病,其发病机制为Ⅳ型胶原α 链基因突变导致的基底膜损伤,临床表现以血尿、进行性肾功能减退,伴有高频感音神经性耳聋和眼部异常为特征[1]。AS 发生率约为1/5 000,在儿童慢性肾功能衰竭中约占3.0%[2]。AS 最主要的遗传方式为X 连锁显性遗传,是由于COL4A5 基因突变所致,约占85%,其他遗传方式为常染色体显性或隐性遗传,由COL4A3 和(或)COL4A4 基因突变引起,约占15%[3]。近年来,随着基因诊断技术的开展,使大部分AS病例在基因水平得到了确诊,但目前国内尚无应用外显子捕获-第二代测序技术来检测儿童AS 基因突变的报道,本文采用该技术对2010 年11 月至2012 年6 月北京军区总医院附属八一儿童医院(我院)符合AS 临床诊断[4]患儿进行相关基因检测,并总结临床特点,以期丰富AS 基因突变数据库,进一步提高对该病的认识。
1 病例资料
1.1 伦理和知情同意 本研究经我院伦理委员会批准,患儿由其父母签署知情同意书同意采血进行基因检测。
1.2 临床特征 表1 显示,4 例临床诊断AS 患儿均为男性,年龄6 ~8 岁,平均7 岁;病程0 ~5 年,平均2.25 年。均表现为镜下或肉眼血尿,尿RBC 形态均提示肾小球性血尿。均表现为不同程度蛋白尿,其中例2 曾达肾病水平蛋白尿。目前4 例肾功能均正常,均进行了肾穿刺活检,光镜和常规免疫荧光检查未见特异病变,电镜检查显示典型的AS 基底膜病变。例1 肾Ⅳ型胶原免疫荧光检查显示:α3链+ + +,α5 + +,例3 皮肤Ⅳ型胶原免疫荧光检查显示α5 +。4 例均接受了纯音测听检测,其中例2 表现为高频听力区受损。4 例均接受眼裂隙灯和眼底检查,例1 右侧视网膜脱色素改变,余3 例正常。经询问病史,4 例患儿家族中均无终末期肾病(ESRD)成员,例2 和3 母亲均有镜下血尿(表1)。
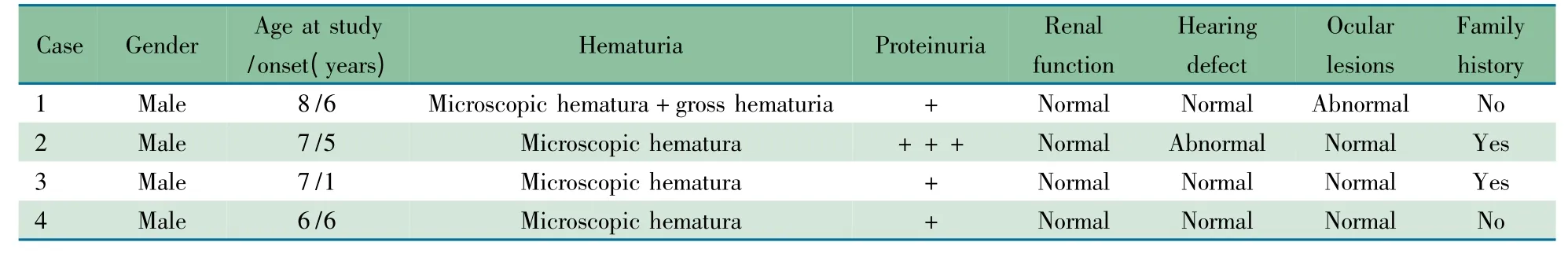
表1 4 例AS 患儿的主要临床资料Tab 1 Major clinical data of 4 patients with AS
1.3 COL4A5、COL4A4 和COL4A3 基因外显子捕获-第二代测序
1.3.1 文库构建 患儿及其母亲采集静脉血,提取基因组DNA(the QIAamp DNA Blood Midi Kit,Qiagen,Hilden,Germany),合格的基因组DNA 被随机打断(Covaris),纯化长度200 ~250 bp 的片段;之后纯化的DNA 利用T4 DNA连接酶、T4 phosphonucleotide kinase 和Escherichia coli DNA聚合酶Klenow 片段进行末端修复,再按照Illumina 的操作说明在片段两端加上A 碱基,最后在两端加上接头,加上接头后的模板进行磁珠纯化。
1.3.2 杂交 已加接头纯化后的模板进行捕获前PCR,PCR 产物纯化后与自主设计的人类基因芯片(Roche NimbleGen,Madison,USA)杂交68 ~72 h,目标片段与磁珠结合被捕获,而未杂交的片段被洗掉,洗脱后的目标片段再进行一次捕获PCR。
1.3. 3 测序 捕获片段PCR 产物利用Agilent 2100 Bioanalyzer 和ABI stepOne 进行富集度检测,通过质控后的PCR 产物在Illumina HiSeq 2000 上进行双向各90 bp 高通量测序,测序数据利用Illumina 的软件进行分析处理。测序重复3 次。
1.4 COL4A5 基因突变结果 突变位点测序reads 比对图发现4 个突变,即Gly132Glu、Gly1238Arg、Gly267Arg 和Gly1033Ser,均为纯合突变。例1 COL4A5 基因第7 外显子第395 位碱基鸟嘌呤(G)突变为腺嘌呤(A),即c.395 G >A,导致GGA→GAA 密码子的改变,使得第132 位氨基酸从甘氨酸(Gly)突变为谷氨酸(Glu),即p.Gly132Glu,其母亲不携带该突变(图1A);例2 COL4A5 基因外显子41 第3712 位碱基鸟嘌呤(G)突变为胞嘧啶(C),即c.3712 G >C,导致GGC→CGC 密码子的改变,使得第1238 位氨基酸从Gly 突变为精氨酸(Arg),即p.Gly1238Arg,其母亲携带该突变(图1B);例3 COL4A5 基因第14 外显子第799 位碱基鸟嘌呤(G)突变为腺嘌呤(A),即c. 799 G >A,导致GGG→AGG 密码子的改变,使得第267 位氨基酸从Gly 突变为Arg,即p.Gly267Arg,其母亲携带该突变(图1C);例4 COL4A5 基因第35 外显子第3097 位碱基鸟嘌呤(G)突变为腺嘌呤(A),即c.3097G >A,导致GGU→AGU 密码子的改变,使得第1033 位氨基酸从Gly 突变为丝氨酸(Ser),即p.Gly1033Ser,其母亲不携带该突变(图1D)。4 例确定为X 连锁遗传型,2 例为新生突变,2 例来源于母亲。SIFT 和PolyPhen 软件进行蛋白功能预测均显示为有害突变。
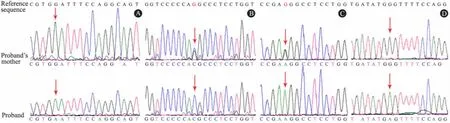
图1 4 例AS 患儿及其母亲COL4A5 基因突变结果Fig 1 Four pathogenic mutations of COL4A5 gene in the 4 families
经检索人类基因突变数据库以上4 种突变均为新突变。4 种突变的千人频率(千人计划中93 个中国人测序样本中关于此SNP 的频率信息)和本地频率(本地收集的>100 个正常人测序样本中关于此SNP 的频率信息)均为0。
2 讨论
AS 共有3 个致病基因,分别为COL4A5(51 个外显子)、COL4A3(52 个外显子)和COL4A4(48 个外显子),外显子数目庞大且无突变热点,采用传统PCR 测序方法检测突变耗费大量的时间和人力。虽有学者通过取患儿皮肤标本培养成纤维细胞应用RT-RCR 测序方法来检测COL4A5基因[5],较快捷,但该方法的缺点是:①取皮肤组织会给患儿带来痛苦;②在某些无义突变介导的mRNA 降解或外显子大片段缺失等情况下可导致突变检测失败。因此,寻找一种简洁、快速的AS 基因突变检测方法,成为了医学研究者努力的目标。外显子捕获-第二代测序技术可在同一张芯片上高特异性和高覆盖率捕获研究者感兴趣的目标外显子区域,利用第二代测序直接解析数据,筛选出突变。该方法只需6 个工作日即可完成,节省人力。因此,采用外显子捕获-第二代测序技术进行AS 基因诊断,比传统的PCR 测序法更为实用[6]。晚近,Artuso 等[7]用该技术对3 例疑似AS 患儿进行了基因分析,发现2 例患儿分别于COL4A3 和COL4A4 基因发生复合杂合突变。目前国内尚未见采用该技术进行AS 研究的报道。
AS 是由于Ⅳ型胶原α 链基因突变导致的基底膜损伤,Ⅳ型胶原是构成肾小球基底膜(GBM)的重要组成部分。Ⅳ型胶原由α1 ~α6 链构成,分别由COL4A1 ~COL4A6 基因编码表达[8]。COL4A5 基因位于Xq22 区域,全长250 kb,共51 个外显子,编码含1 685 个氨基酸的α5(Ⅳ)链,其中间胶原区(1 430 个氨基酸)为富含甘氨酸(Gly-X-Y)的三联结构[9]。甘氨酸侧链体积最小,使得3 个甘氨酸残基足能容进紧密折叠的三螺旋内部,若甘氨酸的位置被一个体积较大的氨基酸替代,异常折叠的三螺旋胶原分子对蛋白酶降解的敏感性明显增加而易于被降解[10]。迄今,已报道400 多种COL4A5 基因突变,尚未见热点突变报道。错义突变占30%,多数为发生于α5(Ⅳ)链的胶原区且为甘氨酸残基的替代[11]。本研究中4 例AS 患儿突变均发生在COL4A5 基因,分别为Gly132Glu、Gly1238Arg、Gly267Arg和Gly1033Ser,均为甘氨酸被替代的错义突变,用SIFT 和PolyPhen 软件进行蛋白功能预测均为有害突变。有研究报道,发生在COL4A5 基因第23 ~51 外显子突变的患者会较早进入肾功能衰竭期,或更容易合并眼、耳的损害,但突变对α5 (Ⅳ)链蛋白功能影响尚待进一步研究[12]。Gly132Glu 和Gly1033Ser 为新生突变,发生可能与以下因素有关:①患儿在发育过程中受物理、化学和生物等因素的诱变作用所致;②患儿母亲如生殖细胞发生新生突变则导致其表现为生殖细胞嵌合体,在受精卵形成过程中携带突变的卵细胞可能会将突变传递给下一代,传递概率由突变型生殖细胞占总生殖细胞的比例来决定[13]。虽然携带新生突变患儿的父母再生育该类患儿可能性很小,但是携带新生突变患者婚后再生育后代仍有遗传风险,因此,新生突变患者婚后生育时有必要进行遗传咨询和产前诊断。
甘氨酸替代类型与临床表型之间并无显著的相关性。如COL4A5 错义突变,导致869 位的甘氨酸被精氨酸替代和α5(Ⅳ)链的1143 位的甘氨酸被门冬酰胺替代的AS 患者,临床表现均较重[14,15]。但某些同样发生于α(Ⅳ)链胶原区的甘氨酸替代却产生较轻的临床表现[16]。还有某些氨基酸替代并不导致Ⅳ型胶原分子的病理改变而出现临床症状[17]。说明被替代甘氨酸的位置或者替代氨基酸本身都可以对三螺旋折叠产生影响,并最终影响其临床表型的严重性。本文4 例患儿均为甘氨酸替代的突变,目前肾功能均正常,临床表现差异不大,但疾病的发展和预后需要进一步随访研究。
据报道,在X 连锁显性遗传型AS,肾脏和皮肤Ⅳ型胶原免疫荧光染色大多数表现为α3(-)、α4(-)和α5(-)。而本文例1 和3 肾脏或皮肤Ⅳ型胶原免疫荧光染色均为(+)。复习文献得知,有15% ~20%AS 患者肾组织α3、α4 及α5 链的分布可以正常[18],如王芳等[19]报道了1 例7 岁AS 男性患儿,COL4A5 基因检测显示g.917G >A突变,而肾脏Ⅳ型胶原免疫荧光检测α3、α4 及α5 链均阳性,皮肤α5 链均阳性,推测某些突变可能对α5 链的结构和功能影响不大,因而不会导致Ⅳ型胶原检测阴性。因此,对于AS 而言,临床表型以及肾脏、皮肤的Ⅳ型胶原染色及电镜检查仅为诊断提供辅助信息,该病的确诊最终依然靠基因诊断。
基因诊断是确诊AS 的最可靠手段。本研究对4 例男性AS 临床诊断患儿进行了基因分析,最后证实突变均发生在COL4A5 基因上,早期基因诊断防止了误诊误治,避免了激素及免疫抑制剂给患儿带来的不良反应。因此,对于疑似AS 患儿,尤其是男性,不论其母亲是否有血尿,均建议首选COL4A5 基因检测以明确诊断。目前关于COL4A5基因突变的功能研究仍是一个难点和热点,本课题组将开始新突变功能研究,在蛋白水平研究基因突变对AS 的影响。
[1]Hudson BG, Tryggvason K, Sundaramoorthy M,et al. Alport's syndrome, Goodpasture's syndrome, and type IV collagen. N Engl J Med,2003,348(25): 2543-2556
[2]Levy M, Feingold J. Estimating prevalence in single-gene kidney diseases progressing to renal failure. Kidney Int ,2000,58(3):925-943
[3]Pirson Y. Making the diagnosis of Alport's syndrome. Kidney Int,1999,56(2): 760-775
[4]Flinter F, Cameron J, Chantler C, et al. Genetics of classic Alport's syndrome. Lancet,1988,2(8618):1005-1007
[5]Inoue Y, Nishio H, Shirakawa T, et al. Detection of mutations in the COL4A5 gene in over 90% of male patients with X-linked Alport's syndrome by RT-PCR and direct sequencing. Am J Kidney Dis,1999,34(5):854-862
[6]Luo DF(罗东凤), Wang SY. Whole-exome sequencing and its applications in the research of hereditary disease. International Journal of Genetics (国际遗传学杂志).2012,35(3):173-177
[7]Artuso R, Fallerini C, Dosa L,et al. Advances in Alport syndrome diagnosis using next-generation sequencing. Eur J Hum Genet,2012,20(1): 50-57
[8]Hudson BG,Tryggvason K, Sundaramoorthy M, et al. Alport's syndrome, Goodpasture's syndrome and type IV collagen. N Engl J Med,2003,348(25):2543-2556
[9]Tryggvason K,Zhou J,Hostikka L,et al.Molecular genetics of Alport syndrom.Kidney Int,1993,43(1):38-44
[10]Fang QY(房秋园),Wang YF,Zhang YK, et al. Proteins structure change of COL4A4 gene point mutation and its association with phenotype in thin basement membrane nephropathy.Chin J Nephrol(中华肾脏病杂志),2010,26(1):3-8
[11]Hertz JM, Juncker I, Persson U, et al. Detection of mutations in the COL4A5 gene by SSCP in X-linked Alport syndrome.Hum Mutat,2001,18(2):141-148
[12]Tan R, Colville D, Wang YY, et al. Alport retinopathy results from "severe" COL4A5 mutations and predicts early renal failure. Clin J Am Soc Nephrol,2010,5(1): 34-38
[13]兰风华,王志红, 柯龙凤,等.新生突变与人类疾病.中国遗传学会第八次代表大会暨学术讨论会论文摘要汇编,2004-2008
[14]Barsotti P, Muda AO, Mazzucco G, et al. Distribution of alpha-chains of type IV collagen in glomerular basement membranes with ultrastructural alterations suggestive of Alport syndrome.Nephrol Dial Transplant,2001,16(5):945-952
[15]Zhou J, Hertz JM, Tryggvason K. Mutation in the alpha 5(IV)collagen chain in juvenile-onset Alport syndrome without hearing loss or ocular lesions: detection by denaturing gradient gel electrophoresis of a PCR product. Am J Hum Genet,1992,50(6):1291-1300
[16]Inoue Y, Nishio H, Shirakawa T, et al. Detection of mutations in the COL4A5 gene in over 90% of male patients with X-linked Alport's syndrome by RT-PCR and direct sequencing. Am J Kidney Dis,1999,34(5):854-862
[17]Wang F(王芳),Ding J,Yu LX, et al. Relationship between genotype and phenotype in Alport syndrome: analysis at α5(Ⅳ) chain mRNA level. Chin J Nephrol(中华肾脏病杂志),2003,19(5):281-285
[18]Gross O, Net zer KO, Lambrecht R,et al.Meta-analysis of genotype-phenotype correlation in X-linked Alport syndrome impact on clinical counseling. Nephrol dial transplant,2002,17(7): 1217-1227
[19]Wang F(王芳),Ding J,Yu LX, et al. COL4A5 mutation in an Alport syndrome boy with normal α(Ⅳ) chains of basement membranes.Chinese Journal of Practical Pediatrics(中国实用儿科杂志),2003,18(6):236-239
猜你喜欢
电子科技大学学报(2022年5期)2022-10-29
中国生殖健康(2020年4期)2021-01-18
中国生殖健康(2018年4期)2018-11-06
国外畜牧学·猪与禽(2018年10期)2018-05-14
国外畜牧学·猪与禽(2018年8期)2018-05-14
国外畜牧学·猪与禽(2018年7期)2018-05-14
渔业研究(2018年5期)2018-01-17
人间(2015年11期)2016-01-09
中国医疗美容(2015年4期)2015-04-27
郑州大学学报(医学版)(2015年2期)2015-02-27
