
氧化石墨负载的手性二胺铑催化剂催化芳香酮的不对称氢转移反应
2013-12-18 06:55:42刘克堂徐圆圆段梦圆魏玢莹陈倩芸程探宇刘国华
上海师范大学学报·自然科学版 2013年1期
刘克堂, 徐圆圆, 段梦圆, 魏玢莹, 陈倩芸, 程探宇, 刘国华
(上海师范大学 生命与环境科学学院,上海 200234)
0 引 言
近年来,手性多相催化反应越来越受到催化工作者的关注[1-2].由于反应容易控制,催化剂易于与反应物分离,循环使用方便,对环境污染少等优点,多相催化引起了科学家广泛研究的兴趣.手性醇可以作为化学医药等领域的重要原料或中间体,而其来源主要是酮的不对称还原反应,并且反应条件温和,因此酮的多相不对称氢转移反应受到了广泛的关注,并取得了一定的研究成果[3-4].
碳材料包括石墨、石墨烯、氧化石墨、碳纳米管、碳纳米纤维等[5-6],石墨是一类重要的化学材料,石墨在使用前通常需要经过氧化方法使其表面功能化,而化学氧化法是制备氧化石墨的主要方法.化学氧化法的过程包括石墨氧化、水解和纯化3个主要阶段,其原理是以强氧化剂氧化石墨,将含氧基团引入到石墨层间形成氧化石墨.石墨的氧化方法主要有Staudenmaier[7]法、Brodie[8]法和Hummers[9]法3种,它们都是用无机强质子酸(如浓硫酸、发烟HNO3或它们的混合物)处理原始石墨,将强酸小分子插入石墨层间,再用强氧化剂(如KMnO4、KClO4等)对其进行氧化.其中Hummers氧化法具有非常突出的优点:安全性高、合成时间短、副产物少等.与Hummers法和Brodie法相比,Staudemaier法使用浓硫酸和发烟硝酸混合酸处理石墨,对石墨层结构的破坏较为严重.
目前功能化氧化石墨被广泛应用于电化学、催化剂固载等领域.在多相催化反应中碳材料一直受到广泛的关注,大量的研究结果表明碳载体结构对催化剂的催化活性有很大影响,氧化石墨具有规整的片状表面结构,表面丰富的羧基、羟基和环氧基等活性基团,有助于进行材料功能化,疏水表面有利于提高有机反应的速度.Scheuermann[10]等把Pd纳米颗粒负载到氧化石墨上得到了负载型Pd催化剂,该催化剂在醇溶液中对Suzuki-Miyaura偶联反应具有较高的催化活性.Lemus-Yegres[11]等把3-氨丙基三甲氧基硅烷及COD-Rh嫁接到氧化石墨上得到非均相催化剂,该催化剂对酮或烯烃的加氢反应具有较高的催化活性.
尽管一些研究已经涉及到,但在不对称催化反应中仍存在空白[12-15],在这个研究中采用Hummers方法合成了氧化石墨材料(GOM),并在氧化石墨片上修饰手性二胺基团,然后与金属铑配位得到非均相催化剂.该催化剂对芳香酮的不对称氢转移反应显示高的催化活性和立体选择活性.
1 实验部分
1.1 试剂与仪器
试剂:[Cp*RhCl2]2上海百灵威公司购得,芳香酮在上海达瑞化学有限公司购得,高锰酸钾、双氧水、甲酸钠、浓硫酸在国药集团化学试剂有限公司购得.仪器:红外光谱是由FTIR( Nicolet,Magna 550)测定;表面形貌通过透射电子显微镜(TEM)( JEOL JEM2010,200kV )测定,X射线光电子能谱分析由(Perkin-Elmer PHI 5000C ESCA)测定;催化反应的转化率(Conversion)及产物的对映体过量(ee)值是通过GC气相色谱仪 [Supelco β-Dex 120 chiral column(30 m×0.25 mm(i.d.),0.25 μm film)]测定.
1.2 (R,R)-TsDPEN(1)的制备
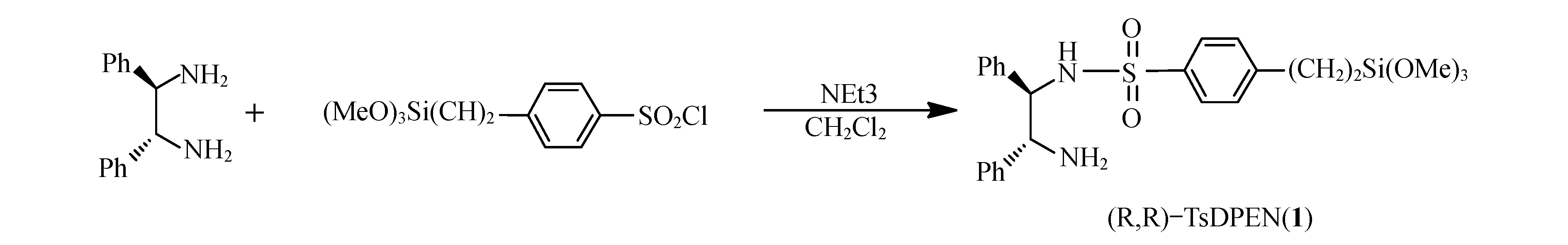
Scheme 1 (R1R)-TSDPENCD的制备
按照文献方法[16],将(R,R)-1,2-二苯基乙二胺(254.7 mg,1.2 mmol)和NEt3(1 mL,7 mmol),CH2Cl2(5 mL)加入圆底烧瓶中,然后在冰浴条件下逐滴加入(324.8 mg,1.0 mmol)2-(4-氯磺酰苯基)乙基三甲氧基硅烷二氯甲烷(10 mL)溶液,移走冰浴室温条件下继续反应3 h.反应结束后将溶剂去除,得到粗产物.采用V(Et3N)∶V(CH3OH)∶VCH2Cl2)= 1∶10∶100过柱分离得纯品.1HNMR(400 MHz,CDCl3)δ = 0.89~0.95(m,2H),1.50(s,2H,NH2),2.65~2.70(m,2H),3.59(s,9H),4.12(d,J = 5.4 Hz,1H),4.37(d,J = 5.4 Hz,1H),6.99~7.34(m,14H);13C NMR(75 MHz,CDCl3)δ = 11.0,28.5,50.6,60.4,63.2,126.5,126.9,127.3,127.4,127.8,128.2,128.3,137.3,139.1,141.3,148.8.
1.3 合成Cp*RhTsDPEN-GOM
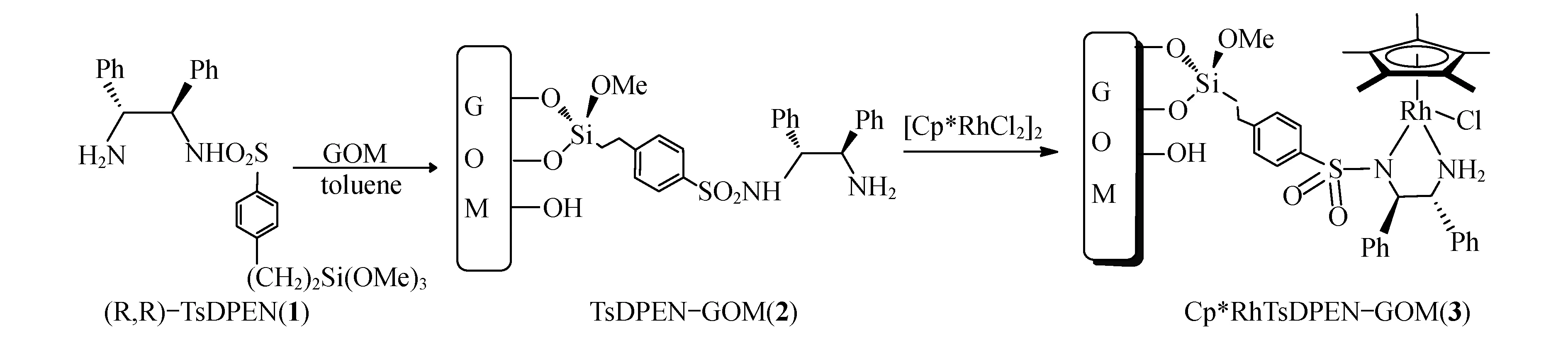
Scheme 2 Cp*RhTsDPEN-GOM
在圆底烧瓶中加入60 mL甲苯和396.5 mg上述制得的(R,R)-TsDPEN(1),再加入1.0 g氧化石墨材料GOM,120 ℃条件下搅拌回流反应24 h;反应完成后过滤分离,并用10 mL甲苯洗涤滤饼, 60 ℃下真空条件下干燥24 h,得黑色固体功能化氧化石墨材料TsDPEN-GOM(2).然后将1.28 g功能化氧化石墨材料TsDPEN-GOM(2)加入到溶有100 mg [Cp*RhCl2]2的60 mL二氯甲烷溶液中,室温条件下搅拌反应6 h,减压蒸除溶剂,得到粗产品索氏提取,固体室温条件下干燥过夜制得氧化石墨负载的手性有机铑金属催化剂Cp*RhTsDPEN-GOM(3).
1.4 催化反应
将Cp*RhTsDPEN-GOM(3)(20.0 mg),HCOONa(104.0 mg),芳香酮(1.0 mmol)和2.0 mL蒸馏水依次加入到10 mL的圆底试管中,40℃条件下搅拌反应5~10 h .反应结束后,向圆底试管中加入2 mL无水乙醚并搅拌,离心(10000 r/min,10 min),有机相过滤分离;乙醚溶液用饱和氯化钠溶液洗涤两次,然后加入无水MgSO4干燥.过滤减压除去乙醚,粗产物再利用小硅胶色谱柱分离处理,得到无色液体即为产物,利用GC气相色谱仪测定其转化率(conv)和对映体过量(ee)值.
2 结果与讨论
2.1 材料催化剂表征
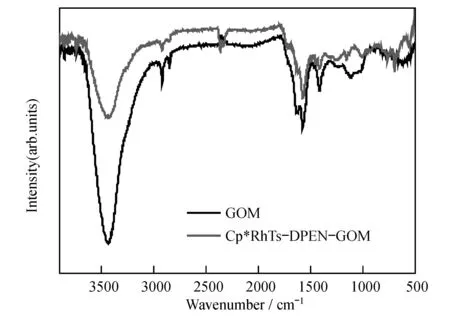
图1 氧化石墨材料及Cp*RhTsDPEN-GOM红外谱图
氧化石墨负载的手性有机铑金属催化剂Cp*RhTsDPEN-GOM(3)通过FTIR、TEM、XPS进行了表征分析.氧化石墨和催化剂的红外光谱如图1所示,图中显示氧化石墨存在一些特征吸收峰:如3429 cm-1羟基伸缩吸收峰,1732 cm-1羰基伸缩吸收峰,1627 cm-1处羟基变形振动吸收峰,另外,1466 cm-1到1657 cm-1显示的是材料中苯环骨架震动的特征吸收峰.对比这些氧化石墨材料特征吸收峰,催化剂存在一些明显特征吸收峰的变化:羟基伸缩振动吸收峰明显减弱,说明部分氧化石墨羟基已经被取代,另外,新出现的1068 cm-1吸收峰归咎于Si-O的特征吸收,这些吸收峰的变化也证实了手性有机金属配合物已成功嫁接到氧化石墨载体上.
图2为透射电子显微镜图像和化学映射,从图2中可以看出已成功制备出面片晶状结构的氧化石墨负载的手性二胺铑催化剂,特别是从化学映射图明显可以看出金属铑均匀分布在氧化石墨表面上(红色部分为金属铑,白色部分为碳).通过上述表征可以进一步说明金属铑成功负载在氧化石墨材料上,并呈现均匀分布,这将有助于提高不对称催化反应的催化活性.

图2 透射电子显微镜(TEM)图像与化学映射
同时,为确定负载型催化剂中有机金属铑配合物的铑的价态和手性微环境状况,又测试了催化剂的X射线光电子能谱.如图3所示,可以清楚地观察到负载型催化剂铑的3d5/2电子能谱类似于均相催化剂的电子能谱,说明了该催化剂具有与均相催化剂相同的手性微环境,这种现象提供了一种探讨不对称催化反应中对应选择性变化的实验证据.
2.2 催化活性
利用合成得到的石墨负载的手性有机铑金属催化剂Cp*RhTsDPEN-GOM(3),考察了多种芳香酮的不对称转移氢化反应,结果列于表1中.按照报道的方法,采用甲酸钠水溶液不对称催化反应体系,以Cp*RhTsDPEN-GOM(3)为催化剂,控制催化剂的摩尔量为底物量的1%,反应温度为40 ℃,另外,由于该反应为固相催化反应,为提高反应转化率,适度地延长了反应时间.从表1中可以看出,石墨负载的手性有机铑金属催化剂Cp*RhTsDPEN-GOM(3)在苯乙酮的不对称氢转移反应中,提供96%的转化率和92%对映选择性(Entry 1).对比原位配位的方法(Entry 2),氧化石墨负载的手性有机铑金属催化剂Cp*RhTsDPEN-GOM(3)在苯乙酮的不对称氢转移反应中显示优于原位配位的催化活性和对映选择活性.
基于苯乙酮的催化效果,考察了系列不同芳香酮的催化性能.由表1可以看出,石墨负载的手性有机铑金属催化剂Cp*RhTsDPEN-GOM(3)对于不同取代的苯乙酮,均呈现出高的催化活性和对映选择活性(Entries 3-7).特别是,苯乙酮的取代基的电性质对该不对称反应没有明显的影响,无论是供电子和吸电子的取代基都呈现出高的催化活性和对映选择活性.
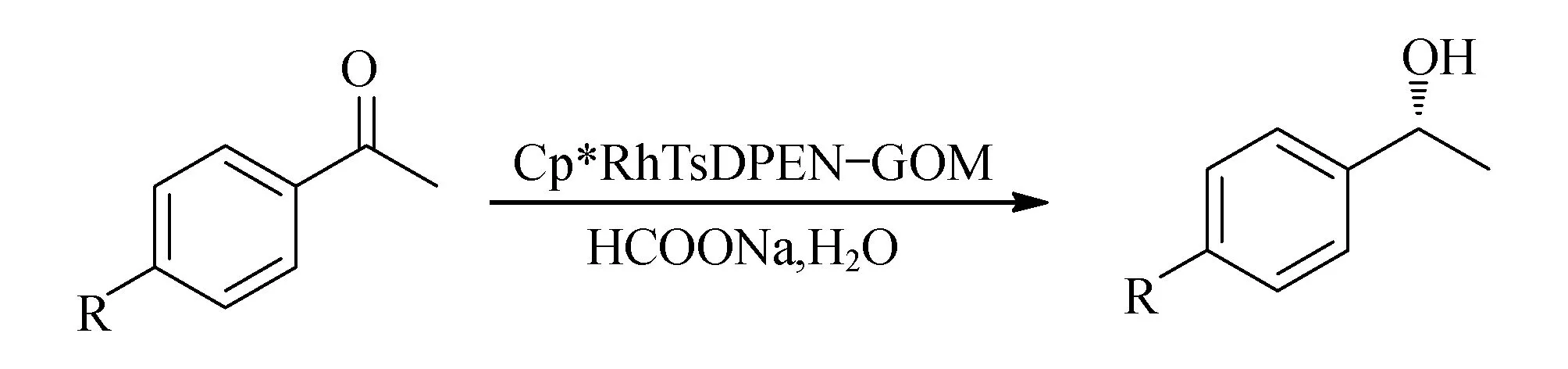
Scheme 3 芳香酮的不对称氢转移反应
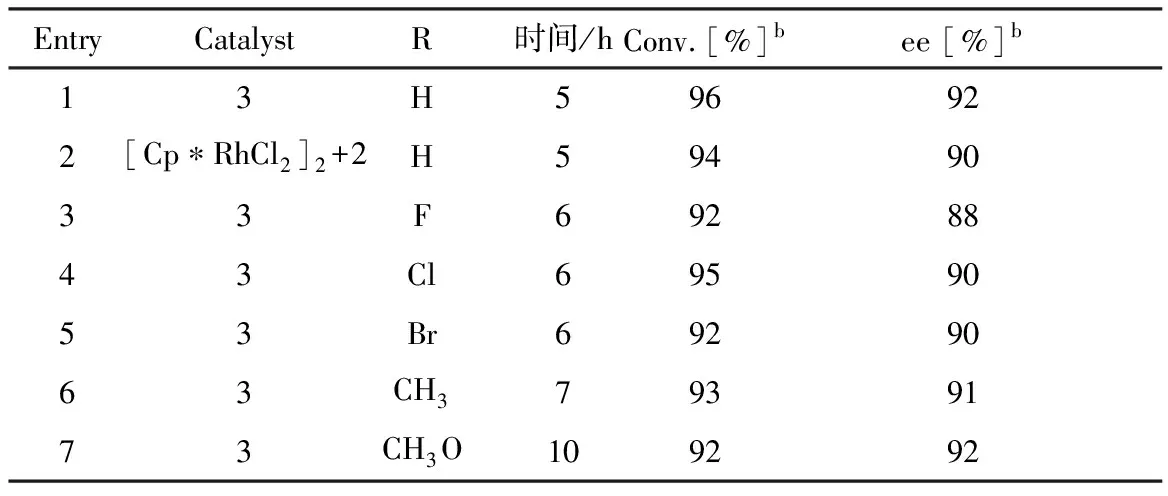
表1 芳香酮的不对称氢转移反应a
反应条件:催化剂3(2 μmol,等离子体发射光谱仪测得)HCDINA(0.68 g,10.0 mmol),芳香酮(0.2 mmol/L)2 mL水反应温度(40 ℃).反应时间(5~10 h).用气相色谱仪分析检测.

图3 X射线光电子能谱分析
3 结 论
本文作者以石墨为原料通过Hummer化学氧化法制备出功能化氧化石墨材料,首次将TsDPEN硅源嫁接到氧化石墨材料上,通过与有机铑配合物配位制备出负载型手性有机金属铑金属催化剂.该催化剂在甲酸钠水溶液催化反应体系中,催化芳香酮的不对称氢转移反应取得了高的催化活性和对映选择活性.
参考文献:
[1] SUN Y Q,LIU G H,GU H Y,et al.Magnetically recoverable SiO2-coated Fe3O4nanoparticles:a new platform for asymmetric transfer hydrogenation of aromatic ketones in aqueous medium[J].Chem Commun,2011,47(9):2583-2585.
[2] LIU G H,WANG J Y,HUANG T Z,et al.Mesoporous silica-supported iridium catalysts for asymmetric hydrogenation reactions[J].J Mater Chem,2010,20(10):1970-1975.
[3] YANG X S,ZHU F X,HUANG J L,et al.Phenyl@Rh(I)-bridged periodic mesoporous organometalsilica with high catalytic efficiency in water-medium organic reactions[J].Chem Mater,2009,21(20):4925-4933.
[4] JIANG D M,GAO J S,YANG Q H,et al.Large-pore mesoporous organosilicas functionalized with trans-(1R,2R)-diaminocyclohexane:synthesis,postmodification,and catalysis[J].Chem Mater,2006,18(26):6012-6018.
[5] GEIM A K,NOVOSELOV K S.The rise of graphene[J].Nature,2007,6(3):183-191.
[6] GOMEZ-NAVARRO C,WEITZ R T,Bittner A M,et al.Electronic transport properties of individual chemically reduced graphene oxide Hummers S,Offeman R.Preparation of graphitic oxide[J].J Am Chem Soc,1958,80(6):1339-1339.
[7] NAKAJIMA T,MATSUO Y.Formation process and structure of graphite oxide[J].Carbon,1994,32(3):469-475.
[8] BRODIE B C.Sur le poids atomique du graphite[J].Ann Chim Phys,1860,59:466-472.
[9] HUMMERS W S,OFFEMAN R E.Preparation of graphite oxide[J].J Am Chem Soc,1958,80(6):1339-1341.
[10] GIL M S,LUI G R,PETER S.Willi bannwarth rolf mu¨lhaupt,palladium nanoparticles on graphite oxide and its functionalized graphene derivatives as highly active catalysts for the suzuki-miyaura coupling reaction[J].J Am Chem Soc,2009,131(23),8262-8270.
[11] LEMUS-YEGRES L J,PÉREZ-CADENAS M,ROMN-MARTNEZ M C,et al.Hybrid rh catalysts prepared with carbon nanotubes of different inner diameter[J].Microporous Mesopotous Mater,2008,109(1-3):305-316.
[12] BAI B Y,YANG H G,WANG P,et al.Enhancement of catalytic performance in asymmetric transfer hydrogenation by microenvironment engineering of the nanocage[J].Chem Commun,2010,46(43):8145-8147.
[13] LIU G H,YAO M,WANG J Y,et al.Enantioselective hydrogenation of aromatic ketones catalyzedby a mesoporous silica-supported iridium catalyst[J].Adv Synth Catal,2008,350(10):1464-1468.
[14] LIU G H,GU H Y,SUN Y Q,et al.Magnetically recoverable nanoparticles:highly efficient catalysts for asymmetric transfer hydrogenation of aromatic ketones in aqueous medium[J].Adv Synth Catal,2011,353(8):1317-1324.
[15] LIU G H,YAO M,ZHANG F,et al.Facile synthesis of a mesoporous silica-supported catalyst for rucatalyzed transfer hydrogenation of ketones[J].Chem Commun,2008(3):347-349.
[16] LIU P N,GU P M,WANG F,et al.Efficient heterogeneous asymmetric transfer hydrogenation of ketones using highly recyclable and accessible silica-immobilized ru-TsDPEN catalysts[J].Org Lett,2004,6(2):169-172.
猜你喜欢
分子催化(2022年1期)2022-11-02 07:10:30
少儿美术(2019年8期)2019-12-14 08:06:58
少儿美术(快乐历史地理)(2018年7期)2018-04-02 19:58:31
岭南音乐(2017年3期)2017-07-18 11:59:40
国外医药(抗生素分册)(2016年4期)2016-07-12 14:25:19
国外医药(抗生素分册)(2016年2期)2016-07-12 14:25:01
通信电源技术(2016年6期)2016-04-20 06:21:22
传奇故事(破茧成蝶)(2015年1期)2015-02-28 09:26:48
郑州大学学报(理学版)(2014年3期)2014-03-01 04:21:06
无机化学学报(2014年12期)2014-02-28 17:33:51
