
三氟乙烷与氟原子氢提取反应的动力学研究
2013-11-21 01:39:30文金淼李永红
化学研究 2013年5期
文金淼,李永红
(河南大学 化学化工学院,河南 开封 475004)
氟利昂(CFCs)又名氟氯烷,是含有氟和氯的有机化合物,它具有优良的化学稳定性,主要用作制冷剂,其次还可以用于清新剂、泡沫灭火剂和发泡剂等. 然而这些物质进入大气后光解产生的Cl 原子可以消耗同温层中的臭氧,臭氧消耗潜值(ODP)高,而且在大气中停留时间长,温室效应潜值(GWP)高[1-3]. 认识到氟利昂对环境的危害和负面影响后,对于如何治理CFCs引起的污染问题并且寻找对环境污染较小的CFCs替代品引起了越来越多研究者的关注[4].
氟利昂(CFCs)的替代品之一是部分氟取代的烷烃化合物(HFCs),这是由于HFCs具有和CFCs相似的物理化学性质. 但是HFCs分子中不含有Cl原子,所以HFCs对臭氧层无污染. 另外,由于HFCs分子中含有C-H键,较易与大气中的原子和自由基反应, 因此HFCs分子比CFCs在大气中存在的时间较短,从而降低了对大气的污染. 1,1,1-三氟乙烷就是HFCs中的一种. CHRISTOPHER等人[5]报道了反应CH3CF3+ F → CH2CF3+ HF在298 ± 4 K温度点的速率常数((2.1 ± 0.8) × 10-12cm3·mol-1·s-1). 然而实验上只给出了一个温度点的速率常数,而对于其他温度区间的速率常数迄今为止还没有理论研究. 本文作者用双水平直接动力学方法[6-8]计算了该反应在200~2 000 K温度范围的速率常数. 首先是在MCG3-MPWPW91//MPW1K/6-311+G(d,p)水平下得到了反应的势能面信息,接着利用变分过渡态理论计算了此反应的速率常数.
1 计算方法
本文涉及的所有电子结构的计算都是在Gaussian03程序下完成的[9]. 用MPW1K方法及6-311+G(d,p)基组对稳定点(包括反应物、过渡态和产物)进行了构型的优化,同时对上述稳定点进行频率计算,通过频率分析来确认所得到的几何构型并得到零点能的数值. 由于过渡态结构对理解反应机理起到至关重要的作用,因此我们从过渡态构型出发,利用内稟反应坐标(IRC)理论,计算得到反应的最小能量途径(MEP),以确定所找到的过渡态确实连接的是反应物和产物. 另外,获得最小能量途径上部分点的一阶和二阶导数,以此来计算反应的曲率和广义的振动频率. 采用MCG3-MPWPW91[10]方法基于MPW1K/6-311+G(d,p) 水平优化得到的所有稳定点以及在最小能量途径上所选的部分点的构型进行单点能量校正,从而得到更为精确的反应能量. 本文的动力学部分的计算是利用Polyrate9.7[11]程序完成的.
2 结果和讨论
2.1 稳定点性质
在MPW1K/6-311+G(d,p)水平下,对所有驻点(包括反应物、过渡态和产物)的构型进行了优化,所得到构型参数以及相应的实验值[12-13]绘于图1. 在此水平下计算得到的CH3CF3和HF的几何构型同实验值符合得很好,最大误差在2.7%以内. 由于反应物CH3CF3是属于Cs点群,因此-CH3基团上具有三个等价的氢原子,并且反应的产物CH2CF3也是Cs点群,所以该反应只存在一个反应通道. 相比反应物CF3CH3中的C-H键,过渡态TS中即将断裂的C-H键的长度增加了4.6%,而与孤立的HF分子中H-F键相比,即将形成的H-F键的键长则拉伸了59%. 很明显,上述过渡态TS属于早垒型过渡态,因此这个反应是早垒型反应. 根据哈蒙德假设[14],早垒型反应应该具有较低的能垒,并且是个放热反应. 这一点将会在下面的讨论中得到验证.

图1 各驻点的几何结构(方括号中为实验值[12-13])长度单位是nm, 角度单位是度Fig.1 Optimized geometries of stationary points. The values in the square bracket are experimental values[12-13]. Bond lengths are in nm and angles are in degrees
表1列出了在MPW1K/6-311+G(d,p)水平下计算得到的所有驻点的振动频率以及其相应的实验值[15-16]. 从表1可以看出计算所得的HF分子的振动频率略小于实验值,其相对偏差仅为3.5%. 不同驻点的性质可以通过对其构型进行振动频率分析来加以确认,也就是反应物和产物的频率全部是实频,而过渡态有且仅有一个虚频. 从表1可以证实这一点,反应物和产物的频率均为实频,而过渡态有且只有一个虚频,计算值为267i.

表1 在MPW1K/6-311+G(d,p)水平下反应物、产物和过渡态的振动频率及相应的实验值(单位: cm-1)Table 1 Calculated frequencies (cm-1) of the reactants, products, and transition-states at the MPW1K/6-311+G(d,p) level
a摘自文献[16].b摘自文献[15]
2.2 反应焓和反应能垒


表2 在MPW1K/6-311+G(d,p)和MCG3-MPWPW91//MPW1K/6-311+G(d,p)水平下计算得到的反应焓,能垒以及相应的实验值(单位:kJ·mol-1)Table 2 Reaction enthalpies and barrier heights (kJ·mol-1) at MPW1K/6-311+G(d,p) and MCG3-MPWPW91//MPW1K/6-311+G(d,p) levels along with the experimental values
a摘自文献[17]
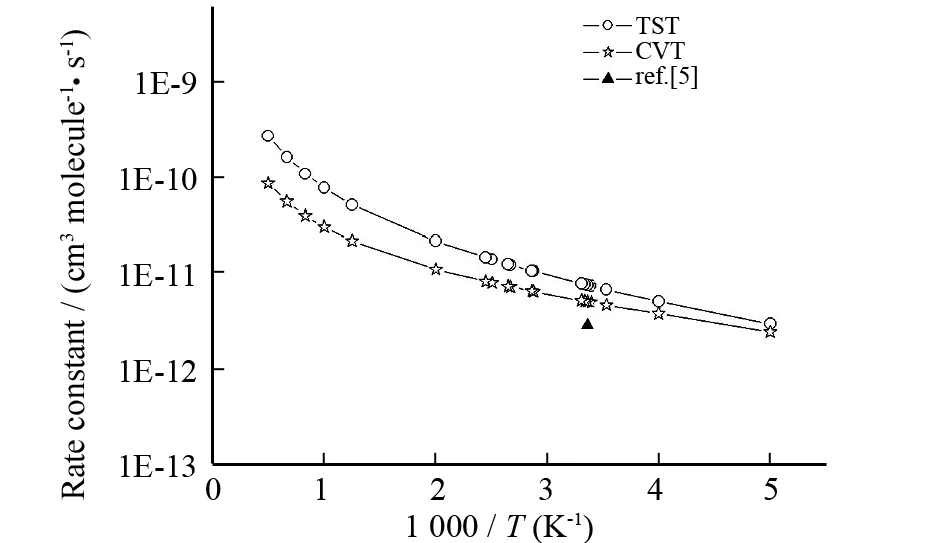
图2 在200~2 000 K温度区间内计算得到的TST和CVT速率常数及实验值随温度变化的曲线Fig.2 Plot of TST and CVT rate constants calculated at the MCG3-MPWPW91//MPW1K level along with the available experimental data versus 1 000/T between 200 and 2 000 K for the title reaction
2.3 动力学计算
在MCG3-MPWPW91//MPW1K/6-311+G(d,p)水平下进行了双水平直接动力学计算,采用变分过渡态理论(VTST)中的传统过渡态理论(TST)和正则变分过渡态理论(CVT) 计算了标题反应在200~2 000 K温度区间的速率常数. 图2绘出了计算得到的TST和CVT的速率常数和实验值与温度的依赖关系. 由图2可知,在整个温度区间内TST的数值大于CVT的数值,说明变分效应在整个温度区间内起着重要的作用. 并且,TST与CVT的差值随着温度的升高而增加,说明变分效应在高温区间比在低温区间的作用明显. 在298 K时,计算所得的CVT的值5.08×10-12cm3·molecule-1·s-1与实验值2.90×10-12cm3·molecule-1·s-1符合得很好.
3 结论
本文运用双水平直接动力学方法研究了CF3CH3与F原子的氢提取反应. 在MCG3-MPWPW91//MPW1K/6-311+G(d,p)水平下,采用变分过渡态理论(VTST)进行了动力学计算. 由于反应物CH3CF3是属于Cs点群,因此-CH3基团上具有三个等价的氢原子,所以该反应只存在一个氢迁移反应通道. 研究结果表明,此氢迁移反应为放热反应. 计算得到的反应焓和实验值符合的很好. 另外,在计算的整个温度区间内TST的数值大于CVT的数值,说明变分效应在整个温度区间内有很重要的作用,在高温区间更为明显. 在298 K,CVT的值5.08×10-12cm3·molecule-1·s-1与实验值2.90×10-12cm3·molecule-1·s-1很相近. 说明在此水平下计算的速率常数是可靠的.
参考文献:
[1] TALHAOUI A, LOUIS F, DEVOLDER P, et al. Rate coefficients of the reactions of chlorine atoms with haloethanes of type CH3CCl3-xFx(x= 0, 1, and 2): experimental and ab initio theoretical studies [J]. J Phys Chem, 1996, 100(32): 13531-13538.
[2] LIU R, HUIE R E, KURYLO M J. Rate constants for the reactions of the hydroxyl radical with some hydrochlorofluorocarbons over the temperature range 270-400 K [J]. J Phys Chem, 1990, 94(8): 3247-3249.
[3] ROWLAND F S. Stratospheric ozone depletion [J]. Annu Rev Phys Chem, 1991, 42: 731-768.
[4] KAISER E W. Relative rate constants for reactions of HFC 152a, 143, 143a, 134a, and HCFC 124 with F or Cl atoms and for CF2CH3, CF2HCH2and CF3CFH radicals with F2, Cl2and O2[J]. J Chem Kinet, 1993, 25(8): 667-680.
[5] MOORE C, SMITH W M. Rate constants for the reactions of fluorine atoms with alkanes and hydrofluorocarbons at room temperature [J]. J Chem Soc Faraday Trans, 1995, 91(18): 3041-3044.
[6] HEIDRICH D. The reaction path in chemistry: current approaches and perspectives [M]. Dordrecht, the Netherlands: Kluwer Academic, 1995: 229.
[7] TRUHLAR D G, GARRENT B C, KLIPPENSTEIN S J. Current status of transition-state theory [J]. J Phys Chem, 1996, 100: 12771-12779.
[8] HU Wei Ping, TRUHLAR D G. Factors affecting competitive ion-molecule reations ClO+C2H5Cl and C2D5ClviaE2and SN2channels [J]. J Am Chem Soc, 1996, 118: 860-869.
[9] FRISCH M J, TRUCK G W, SCHLEGEL H B, et al. Gaussian 03[CP]. Revision A.1: Guassian, Inc., Pittsburgh, PA. 2003.
[10] ZHAO Yan, LYNCH B J, TRUHLAR D G. Multi-coefficient extrapolated density functional theory for thermochemistry and thermochemical kinetics [J]. Phys Chem Chem Phys, 2005, 7: 43-52.
[11] CORCHADO J C, CHUANG Y Y, PAST P L, et al. POLYRATE, version 9.7[CP]. University of Minnesota, Minneapolis. 2007.
[12] MASON M G, VONHOLLE W G, ROBINSON D W. Mid- and far- infrared spectra of HF and DF in rare-gas matrices [J]. J Phys Chem A, 1971, 54: 3491-3499.
[13] LIDE D R. CRC Handbook of chemistry and physics [M]. Boca Raton: CRC Press, 80thed., 1999-2000.
[14] HAMMOND G S. A correlation of reaction rate [J]. J Am Chem Soc, 1955, 77(2): 334-338.
[15] WEBB D U, RAO K N. Infrared spectra and configurations of alkylurea derivatives: normal vibrations on N,N-dimethyl- and tetramethylurea [J]. J Mol Spectrosc, 1968, 28(4): 526-535.
[16] LAFON B, NIELSEN R. Normal coordinate analysis of CF3CH3and CF3CD3[J]. J Mol Spectrosc, 1966, 21: 175-182.
[17] DEMORE W B, SANDER S P, GOLDEN S P, et al. Chemical kinetics and photochemical data for use in stratospheric modeling [M]. California: Jet Propulsion Laboratory Publication, 1997.
猜你喜欢
北京航空航天大学学报(2022年5期)2022-06-06 09:27:18
大学化学(2021年8期)2021-09-26 10:51:16
数学杂志(2020年3期)2020-07-25 01:39:30
数学物理学报(2019年6期)2020-01-13 06:08:18
电脑知识与技术(2018年3期)2018-03-21 09:27:04
数学物理学报(2017年6期)2018-01-22 02:26:49
中学化学(2017年5期)2017-07-07 08:40:47
哈尔滨理工大学学报(2017年1期)2017-04-08 04:16:24
数学物理学报(2016年3期)2016-12-01 05:36:30
中学化学(2016年4期)2016-05-30 16:20:37
