
MALDI-TOF质谱法检测人血清中沙林-丁酰胆碱酯酶加合物
2013-10-25 06:32范崇旭刘尚义代先东温晓波陈冀胜
质谱学报 2013年3期
姚 戈,曹 瑛,范崇旭,刘尚义,代先东,温晓波,陈冀胜
(北京药物化学研究所,北京 102205)
MALDI-TOF质谱法检测人血清中沙林-丁酰胆碱酯酶加合物
姚 戈,曹 瑛,范崇旭,刘尚义,代先东,温晓波,陈冀胜
(北京药物化学研究所,北京 102205)
对生物医学样品进行化学毒剂分析检测是禁止化学武器组织(OPCW)核查小组在毒剂指称使用调查中收集事实的方法之一。丁酰胆碱酯酶(BChE)作为有机磷毒剂在体内的作用靶点,是有机磷毒剂染毒检测的最佳生物指示物之一。利用亲和固相萃取(SPE)技术进一步发展了样品预处理方法,建立了血液样品中染毒BChE的分析方法;利用胰蛋白酶酶解方法,采用基质辅助激光解吸/电离飞行时间质谱(MALDI-TOF MS)比较了血液中BChE在沙林染毒前后的肽指纹谱变化。该方法灵敏度高、快速简便,可用于OPCW生物医学样品中毒剂暴露染毒的追溯性检测。
生物医学样品;有机磷毒剂;丁酰胆碱酯酶;基质辅助激光解吸/电离飞行时间质谱(MALDI-TOF MS)
有机磷毒剂是禁止化学武器组织(OPCW)宣布的毒性最强的致死性化学战剂,已被证实曾在两伊战争及日本东京地铁恐怖袭击事件中使用[1-2]。OPCW一直将该类毒剂作为化学武器核查的重点对象之一,目前已经建立了标准的样品处理及分析方法,但多适用于环境化学样品(土样、水样等)的分析。相较于环境样品,针对生物基质样品(尿样、血样等,亦称生物医学样品)中有机磷毒剂的分析检测则具有更强的溯源性。因此,近年来OPCW开始注重对该类样品分析方法的研究[3]。
有机磷毒剂在体内的作用靶点主要为乙酰胆碱酯酶(AChE)与丁酰胆碱酯酶(BChE),是胆碱酯酶不可逆抑制剂。由于血液中BChE含量远高于AChE,因此在血液样品中,BChE可以看作是有机磷毒剂染毒检测的最佳生物指示物之一[4]。BChE作为有机磷毒剂的生物作用靶点,具有敏感性高、形成加合物半衰期长的特点(8~15天)[5]。目前对BChE加合物的分析多采用液相色谱-串联质谱(LC-MS/MS)或基质辅助激光解吸/电离飞行时间质谱(MALDI-TOF MS)[6-8]。但由于在血液中BChE含量较低(约80 nmol)[5],许多丰度较高的蛋白质(如血清白蛋白、免疫球蛋白)会对BChE的检测产生很大影响。因此在生物医学样品分析中高效的样品预处理技术是分析检测过程的重要因素。
本工作拟以标志性有机磷毒剂沙林为代表,建立沙林-BChE加合物的分析检测方法。并利用BChE与盐酸普鲁卡因酰胺间的亲和特性,结合固相萃取(SPE)技术,建立快速的样品预处理过程,实现对微量血样中沙林-BChE加合物的快速检测,为OPCW生物医学样品有机磷毒剂追溯性检测提供分析方法。
1 试验部分
1.1仪器与试剂
UltraFlex II型 MALDI串联质谱仪:美国Bruker Daltonics公司产品;YM10 超滤管:美国Milipore公司产品;KTA prime液相层析仪:美国Amersham Bioscience公司产品;PowerPac 300 电泳仪:美国BIO-RAD公司产品;ALPHA 2-4 Christ冷冻干燥机:德国Martin Christ公司产品。
1.2主要材料与试剂
人血清:购自北京市红十字会血液中心;沙林,维埃克斯(VX),梭曼:由国家履约实验室馈赠;盐酸普鲁卡因酰胺:美国Aldrich公司产品;溴化氰活化Sepharose 4B:美国Amersham Bioscience公司产品;胰蛋白酶(测序级):美国Roche公司产品;α-氰基-4-羟基肉桂酸(CCA):美国Bruker Daltonics公司产品;硫酸铵(分析纯):北京化学试剂公司产品;其他化学试剂均为分析纯。
1.3试验方法
1.3.1普鲁卡因酰胺-Sepharose 4B亲和凝胶的制备 普鲁卡因酰胺与Sepharose 4B的偶联按文献[9]进行:溴化氰活化的Sepharose 4B冻干粉用1 mmol盐酸溶胀冲洗,充分溶胀的凝胶转移至含有ε-氨基己酸的偶联缓冲液中,于4 ℃搅拌混匀48 h。已偶联的ε-氨基己酸凝胶悬浮于水中,加入盐酸普鲁卡因酰胺至浓度100 mmol/L,然后加入偶联剂EDC,持续搅拌混匀24 h(pH 4.5)。未结合的普鲁卡因酰胺用去离子水洗净,制备好的凝胶用50 mmol 聚丁二酸丁二醇酯(PBS)(pH 8.0)悬浮,置于4 ℃保存。
1.3.2人血清中BChE的分离纯化 10 mL人血清经硫酸铵分级沉淀后,所得上清液用去离子水透析12 h。取制备好的普鲁卡因酰胺-Sepharose 4B亲和凝胶装柱,上样后先用缓冲液A(10 mmol/L PBS,pH 8.0,1 mmol/L EDTA,0.2 mol/L NaCl)和缓冲液B(10 mmol/L PBS,pH 7.4,1 mmol/L EDTA)淋洗,然后用含有0.2 mol/L盐酸普鲁卡因酰胺的缓冲液B洗脱并收集洗脱组分,得到BChE,经充分透析后冷冻干燥保存。SDS-聚丙烯酰胺凝胶电脉(SDS-PAGE)检验纯化结果。
1.3.3BChE的有机磷抑制反应 将BChE(或血清)溶于0.1 mol/L磷酸缓冲液(pH 7.0)中,加入沙林至浓度为1.8 mmol/L,37 ℃温育2 h。过量的沙林透析去除,并对终产物进行酶活测定,验证BChE被完全抑制。
1.3.4BChE(或沙林-BChE)的胶内酶切 配制10%聚丙烯酰胺凝胶进行SDS-PAGE电泳,沙林处理过的BChE样品与BChE空白样品分二孔点样,120 V电泳2 h。目标条带处切取胶块后,经脱色、脱水,加入胰蛋白酶,37 ℃酶解过夜。
1.3.5质谱检测分析 取1 μL酶解原液,与等体积基质CCA饱和溶液混匀,点样于靶板上,空气中自然干燥后形成共结晶,进行质谱分析。所用仪器为Ultraflex MALDI-TOF-TOF质谱仪,采用反射正离子模式。首先用TOF MS模式检测酶解肽段[M+H]+峰,然后用LIFT模式对目标肽段进行串联质谱(MS/MS)分析。采集的质谱数据经Flexanalysis 2.4和Biotools 3.0处理,并链接MASCOT在线数据库进行数据比对及解析。
1.3.6染毒血清样品中有机磷毒剂追溯性检测 取普鲁卡因酰胺-Sepharose 4B亲和凝胶装填空SPE柱(凝胶体积约1 mL),4 ℃存放备用。染毒血清首先用30%硫酸铵分级沉淀,上清液转入YM-10超滤管中超滤去除盐成分,含BChE组分溶于水中。所得样品经自制的普鲁卡因酰胺-Sepharose 4B SPE柱富集后,先用缓冲液A(10 mmol/L PBS,pH 8.0,1 mmol/L EDTA,0.2 mol/L NaCl)和缓冲液B(10 mmol/L PBS,pH 7.4,1 mmol/L EDTA)淋洗,然后用含有0.2 mol/L盐酸普鲁卡因酰胺的缓冲液B洗脱BChE加合物,收集洗脱组分,转入YM-10超滤管中,超滤去除盐成分,含BChE组分溶于20 μL水中存放。取4 μL样品,加热变性3 min(100 ℃),冷却后加入胰蛋白酶,37 ℃酶解2 h。取1 μL酶解液进行质谱分析,SPE柱再生后用去离子水平衡后可重复使用。
2 结果与讨论
沙林进入血液作用于BChE,与活性位点Ser198发生亲核反应,形成BChE加合物,从而导致酶失活,引发中毒反应。沙林与BChE结合后,会逐渐发生水解反应,引起进一步的衰变,即所谓的“老化”现象,得到较稳定的老化产物[10-11],示于图1。因此可以通过检测BChE的Ser198位点是否被修饰,来判断生物体是否曾暴露于毒剂环境中[12]。

图1 BChE的沙林染毒反应Fig.1 Covalent binding of sarin to human BChE
2.1BChE的分离纯化
由于血清样品成分复杂,BChE丰度低,因此许多高丰度蛋白质在酶解、质谱分析过程中都会产生干扰[13-14]。首先利用普鲁卡因酰胺-Sepharose 4B亲和层析色谱对血清中的BChE进行纯化。血清经过硫酸铵沉淀、亲和层析色谱处理后,其中的BChE已达到一定的纯度,可用于肽指纹谱的建立,示于图2。实验表明,该方法对沙林-BChE加合物的富集与纯化有效,也为建立快速的预处理方法提供依据和参考。

a.硫酸铵沉淀所得样品;b.亲和层析所得样品图2 BChE纯化样品SDS-PAGE电泳图Fig.2 SDS-PAGE electropherogram of purified BChE
2.2BChE染毒前后肽指纹谱比较
纯化后的BChE用胰酶酶解后进行MALDI-TOF质谱分析,获得了未染毒BChE样品肽指纹谱,示于图3a,此谱图在MASCOT数据库中的搜索比对结果与人血清BChE高度一致(匹配值154,误差率8.1×1012),BChE序列的覆盖率为25%(人血清BChE登记号P06276)。其中[M+H]+2 928.538 u所对应的是含有活性位点Ser198肽片段(191SVTLFGES198AGAASVSLHLLSPGSHSL-FTR219)。用同样的方法也获得了沙林染毒后BChE肽指纹谱,示于图3b。
BChE被沙林染毒后,[M+H]+2 928.538 u离子峰消失,出现[M+H]+3 048.411 u离子峰,示于图3。这与沙林-BChE加合物的修饰位点在Ser198的推测相符(修饰肽段[M+H]+理论值为3 048.6 u)[11]。同时也检测到加合物的老化产物,分子离子峰为 3 006.456 u。
为进一步确认沙林的被修饰位点,将染毒前后含Ser198的目标肽段作为母离子进行串联质谱分析。未染毒肽片段([M+H]+2 928.538 u)和染毒后肽片段([M+H]+3 048.411 u),分别示于图4a和4b。谱图中丰度较高的碎片离子大部分为y系列离子,丰度最高的[M+H]+对应的是Ser210与Pro211间断裂产生的y9碎片离子(PGSHSLFTR),这与脯氨酸N端极易碰撞诱导解离的报道相符[15]。
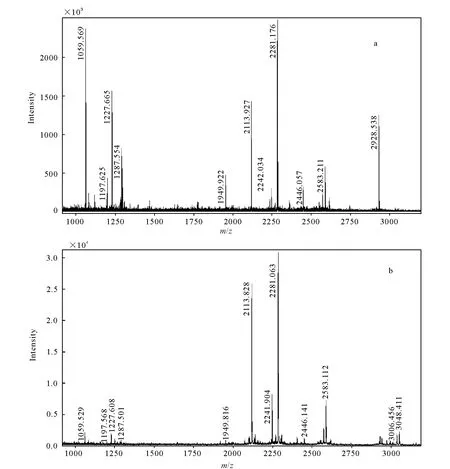
图3 BChE染毒前(a)和染毒后(b)MALDI-TOF肽指纹谱图Fig.3 MALDI-TOF peptide mass fingerprint of before exposure (a) and after exposure (b) for BChE
根据y系列离子对谱图进行手工解析,从图4a可得到包括活性位点Ser198在内的21个连续的氨基酸残基序列194LFGESAGAASVSLHLLSPGSH214,从图4b可得到的连续序列为198S*AGAASVSLHLLS××SH214。比较碎片离子峰,y22(SAGAASVSLHLLSPGSHSLFTR)在染毒前后发生了变化,图4a中y22 [M+H]+为2 194.948 u,图4b中y22 [M+H]+为2 314.839 u,相对分子质量增加了120 u,恰好是沙林磷酰化基团的修饰,因此直接证明了沙林对BChE Ser198的修饰。而其他位置的Ser(如Ser203、Ser205、Ser210、Ser213)对应的y17、y15、y10、y7离子与空白样品相比,相对分子质量均未发生变化,该结果证明了沙林可专一作用于BChE的Ser198位点。
图4b中丰度最高的是[M+H]+2 910.476 u,与母离子质荷比相差约138 u,这可能是由于肽段在二级碎裂过程中,在高能量作用下,磷酰化基团从活性位点Ser198解离下来,Ser198进一步脱水形成碎片离子。但是与空白样品相比,未发现被修饰后y23、y24、y25、y26碎片离子,这可能是碎片离子存在磷酰化基团导致质谱信号变弱所致。从沙林染毒后的BChE肽指纹谱中也能观察到此现象,即磷酰化的肽段离子峰强度大大减弱。因此增加磷酰化肽段的质谱响应信号,将是提高方法灵敏度的重要手段。
为了在实际样品分析时可以对谱图做出快速判断,经过比较分析,给出了BChE的常见特征肽段,列于表1。
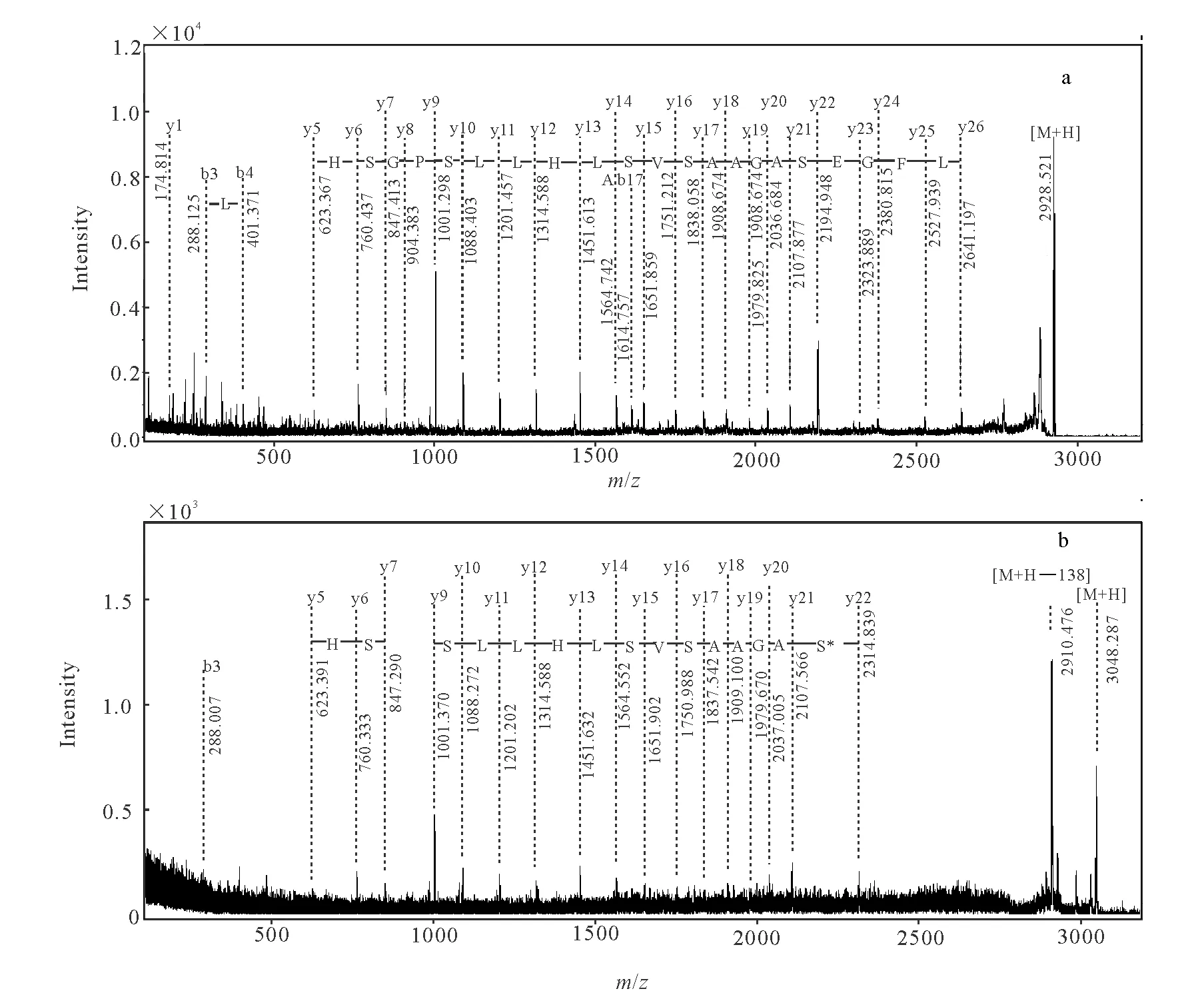
图4 染毒前(a)和染毒后(b)BChE191~209的MS/MS谱图Fig.4 MS/MS spectra of before exposure (a) and after exposure (b) for tryptic peptide BChE191—209
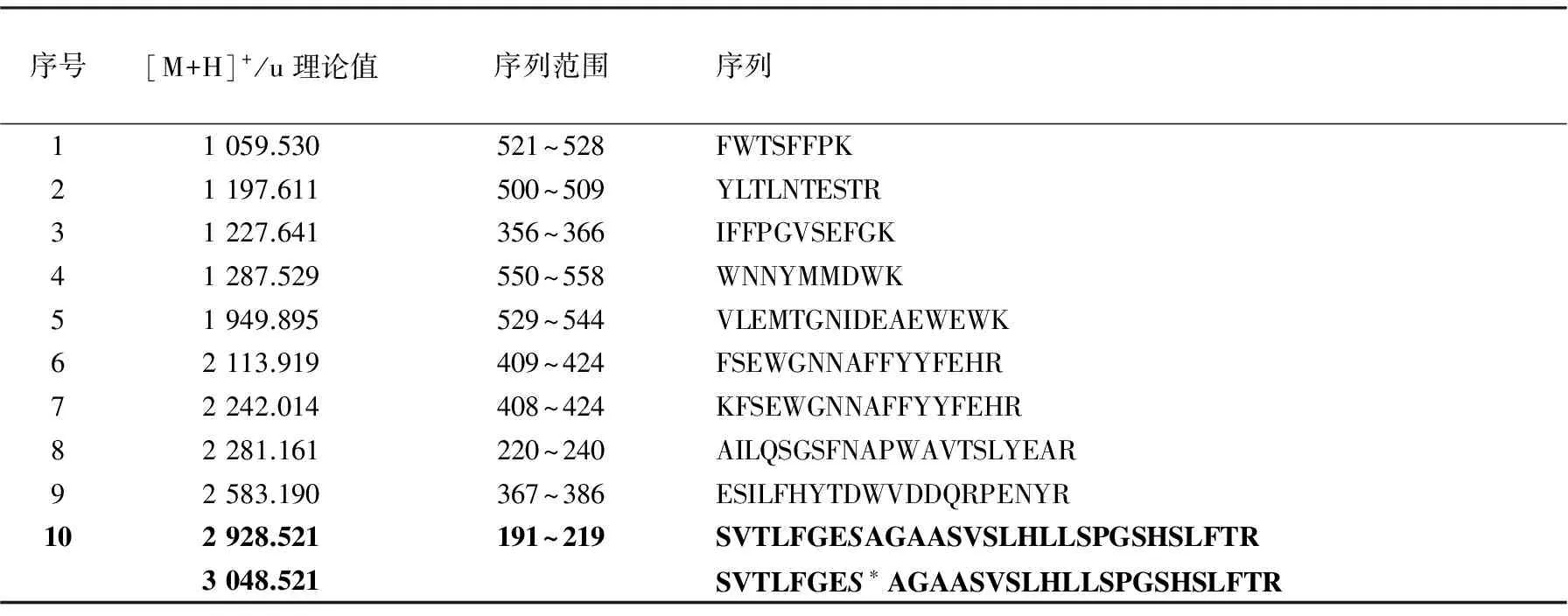
表1 BChE特征性指纹片段
注:粗体表示含有活性位点Ser198的目标肽段,上列为未染毒肽段,下列为沙林染毒后肽段;S表示活性位点Ser198,S*表示被沙林修饰的Ser198
2.3血液样品中沙林-BChE加合物检测及在有机磷毒剂染毒中的应用
由于血清样品中BChE丰度低,因此样品的亲和预处理方法是整个分析过程中重要的一步。在实际样品分析时,一般染毒样品量较少,常规的亲和色谱会由于非特异性吸附等原因损失目标物,导致错误的分析结果。本研究利用亲和层析原理,采用SPE作为预处理技术,尝试对1 mL染毒血清样品中的BChE进行检测。
对血清样品进行分级沉淀处理后,首先用自制的普鲁卡因酰胺-Sephrose 4B SPE柱对血清进行预处理,洗脱液经超滤除盐后,按1.3对样品进行酶解、质谱分析,其肽指纹谱图示于图5。可以看到,虽然使用快速分离技术导致使用肽指纹谱分析的样品纯度不高,但仍可以很清晰地归属表1中的9个BChE胰酶解特征片段,表明样品中存在BChE,根据[M+H]+为3 048.526 u可进一步断定沙林-BChE加合物的存在。

图5 微量染毒血样中BChE的MALDI-TOF肽指纹谱图Fig.5 MALDI-TOF spectra of tryptic peptides of BChE in microscale serum exposure to sarin
由于肽指纹谱是根据酶解片段相对分子质量的变化来判定BChE是否被染毒,因此该方法还应用于其他有机磷毒剂染毒的判断[16-17]。利用所建立的快速样品预处理技术,对其他有机磷毒素染毒血清样品进行了追溯性检测,均达到了预期的检测目的,列于表2。

表2 血清样品中有机磷毒剂染毒检测MS结果
注:粗体表示各种有机磷毒剂染毒后目标肽段相对分子质量
3 结论
本研究利用胰蛋白酶酶解方法[18],结合MALDI-TOF质谱,进一步发展了样品预处理方法,建立了血液样品中染毒BChE的鉴定方法。该方法样品用量少,操作简便、省时,灵敏度高,可用于OPCW生物医学样品的毒剂暴露染毒追溯性检测。同时,由于不同毒剂与BChE活性位点结合的磷酰化基团不同,还可以通过目标肽段相对分子质量的变化来鉴定毒剂的种类。利用该原理及方法还可以进行有机磷农药及氨基甲酸酯类杀虫剂中毒的血清检测[19-20],适用于农药中毒检测等领域。
[1] Report of the specialists appointed by the secretary-general to investigate allegations by the Islamic Republic of Iran concerning the use of chemical weapons. UN document S/16, 433 of 26 March 1984.
[2] OKUMURA T, TAKASU N, ISHIMATSU S, et al.Report on 640 victims of the Tokyo subway sarin attack[J]. Ann Emerg Med, 1996, 28(2): 129-135.
[3] Van der SCHANS M J, FIDDER A, Van OEVEREN D, et al. Verification of exposure to cholinesterase inhibitors: Generic detection of OPCW Schedule 1 nerve agent adducts to human butyrylcholinesterase[J]. J Anal Toxicol, 2008, 32(1): 125-130.
[4] BLACK R M, READ R W. Biological markers of exposure to organophosphorus nerve agents[J]. Arch Toxicol, 2013, 87(3): 421-437.
[5] HOLLAND K E, SOLANO M I, JOHNSON R C, et al. Modifications to the organophosphorus nerve agent-protein adduct refluoridation method for retrospective analysis of nerve agent exposures[J]. J Anal Toxicol, 2008, 32: 116-124.
[6] FIDDER A, HULST A G, NOORT D, et al. Re-trospective detection of exposure to organophosphorus anti-cholinesterases: Mass spectrometric analysis of phosphylated human butyrylcholinesterase[J]. Chem Res Toxicol, 2002, 15: 582-590.
[7] DOORN J A, SCHALL M, GAGE D A, et al. Identification of butyrylcholinesterase adducts after inhibition with isomalathion using mass spectrometry: Difference in mechanism between (1R)- and (1S)-stereoisomers[J]. Toxicol Appl Pharmacol, 2001, 176: 73-80.
[8] LI H, RICORDEL I, TONG L, et al. Ected by mass spectrometry of butyrylcholinesterase adduct in human serum [J]. J Anal Toxicol, 2009, 29(2): 149-155.
[9] GRUNWALD J, MARCUS D, PAPIER Y, et al. Large-scale purification and long-term stability of human butyrylcholinesterase: A potential bioscavenger drug[J]. J Biochem Biophys Methods, 1997, 34: 123-135.
[10] MASSON P, LOCKRIDGE O. Butyrylcholinesterase for protection from organophosphorus poisons; catalytic complexities and hysteretic behavior[J]. Arch Biochem Biophys, 2010, 494(2): 107.doi:10.1016/j. abb.2009.12.005.
[11] Li H, SCHOPFER L M, NACHON F, et al. Aging pathways for organophosphate-inhibited human butyrylcholinesterase, including novel pathways for isomalathion, resolved by mass spectrometry[J]. Toxicol Sci, 2007, 100: 136-145.
[12] ARTURSSON E, ANDERSSON P O, AKFUR C, et al. Catalytic-site conformational equilibrium in nerve-agent adducts of acetylcholinesterase: Possible implications for the HI-6 antidote substrate specificity[J]. Biochem Pharmacol, 2013, 85(9): 1 389-1 397.
[13] ARYAL U K, LIN C T, KIM J S, et al. Identification of phosphorylated butyrylcholinesterase in human plasma using immunoaffinity purification and mass spectrometry[J]. Anal Chim Acta, 2012, 723: 68-75.
[14] SPORTY J L S, LEMIRE S W, JAKUBOWSKI E M, et al. Immunomagnetic separation and quantification of butyrylcholinesterase nerve agent adducts in human serum[J]. Anal Chem, 2010, 82(15): 6 593-6 600.
[15] BRECI L A, TABB D L, YATES J R, et al. CleavageN-terminal to proline: Analysis of a database of peptide tandem mass spectra[J]. Anal Chem, 2003, 75: 1 963-1 971.
[16] JIANG W, CASHMAN J R, NACHON F, et al.Mass spectrometry method to identify aging pathways of Sp- and Rp-tabun adducts on human butyrylcholinesterase based on the acid labile P-N bond[J]. Toxicol Sci, 2013, 132(2): 390-398.
[17] GILLEY C, MACDONALD M, NACHON F, et al.Nerve agent analogues that produce authentic soman, sarin, tabun, and cyclohexyl methylphosphonate-modified human butyrylcholinesterase[J]. Chem Res Toxicol, 2009, 22(10): 1 680-1 688.
[18] MARSILLACH J, HSIEH E J, RICHTER R J, et al. Proteomic analysis of adducted butyrylcholinesterase for biomonitoring organophosphorus exposures[J]. Chem-Biol Interact, 2013, 203(1): 85-90.
[19] Van der SCHANS M J, HULST A G, Van OE-VEREN D, et al. New tools in diagnosis and biomonitoring of intoxications with organophosphorothioates: Case studies with chlorpyrifos and diazinon[J]. Chem-Biol Interact, 2013, 201(1): 96-102.
[20] LI B, RICORDEL I, SCHOPFER L M, et al. Dichlorvos, chlorpyrifos oxon and Aldicarb adducts of butyrylcholinesterase, detected by mass spectrometry in human plasma following deliberate overdose[J]. J Anal Toxicol, 2010, 30(6): 559-565.
DetectionofSarin-ButyrylcholinesteraseAdductinHumanSerumbyMALDI-TOFMassSpectrometry
YAO Ge, CAO Ying, FAN Chong-xu, LIU Shang-yi, DAI Xian-dong, WEN Xiao-bo, CHEN Ji-sheng
(ResearchInstituteofPharmaceuticalChemistry,Beijing102205,China)
The analysis of biomedical samples is one part of the verification programs of OPCW (Organization for the Prohibition of Chemical Weapons), and the results would be used as evidence in case of allegations of chemical agents use. The organophosphorus nerve agents could covalently conjugate with human butyrylcholinesterase (BChE), which is one of the most important biomarkers to verify an exposure to nerve agents. In this paper, the affinity SPE technology was employed as pretreatment method on the case of the detection of serum samples. The difference of peptide mass fingerprints between BChE and sarin-BChE was compared by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS) combined with trypsin digest, and the modified tryptic peptide by sarin ([M+H]+3 048.411 u) was found. The unmodified and modified fragments were also verified by tandem mass spectrometry, and the modified site could be attributed to Ser198. The method of verification of human plasma exposure to sarin can be used to track other organophosphorus agents successfully.
biomedical samples; organophosphorus nerve agents; butyrylcholinesterase; matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS)
O 657.63
A
1004-2997(2013)03-0129-08
10.7538/zpxb.2013.34.03.0129
2013-03-04;
2013-04-09
姚 戈(1983~),男,山东阳信人,博士研究生,军事化学与烟火技术专业。E-mail: bzyaoge@yahoo.com.cn
曹 瑛(1970~),女,山东济宁人,副研究员,从事天然产物化学研究。E-mail: cao_ying2002@hotmail.com
猜你喜欢
河北医学(2020年1期)2020-01-18
兵团工运(2019年1期)2019-12-15
环球时报(2018-04-16)2018-04-16
东西南北(2016年14期)2016-08-16
中国中医急症(2014年4期)2014-05-07
中国医药指南(2014年7期)2014-01-24
药学实践杂志(2011年2期)2011-11-22
军事历史(1993年2期)1993-01-18
