
液相色谱-串联质谱法以锂加合离子定量分析大鼠血浆中紫杉醇和羟基代谢物
2013-10-25 06:32:20范亚新陈笑艳马智宇高志伟钟大放
质谱学报 2013年3期
范亚新,陈笑艳,马智宇,高志伟,钟大放
(1.浙江工业大学,浙江 杭州 310014;2.中国科学院上海药物研究所,上海 201203)
液相色谱-串联质谱法以锂加合离子定量分析大鼠血浆中紫杉醇和羟基代谢物
范亚新1,2,陈笑艳2,马智宇2,高志伟2,钟大放1,2
(1.浙江工业大学,浙江 杭州 310014;2.中国科学院上海药物研究所,上海 201203)
为了提高大鼠血浆中紫杉醇和羟基代谢物的检测灵敏度,本工作探索了加入金属离子的液相色谱-串联质谱联用方法。紫杉醇质谱分析时,易产生碱金属 (Li+、Na+、K+) 加合离子。采用未添加碱金属离子的流动相分析时,[M+H]+质谱响应低,[M+Na]+稳定性差。本实验以碳酸锂作为试剂,同时定量测定大鼠血浆中紫杉醇和羟基代谢物,紫杉醇的线性范围为1.00~8 000 μg/L,3’-p-羟基紫杉醇的线性范围为0.50~50 μg/L。本方法专属性好,准确、快速,适用于注射用紫杉醇 (白蛋白结合型) 的药代动力学研究。锂加合离子稳定性好、质谱响应高、碰撞能量低,适用于一些中性化合物的定量和结构分析。
锂加合离子;紫杉醇;羟基代谢物;液相色谱-串联质谱
紫杉醇是从红豆杉的树皮中提取的天然抗癌化合物,临床应用于多种癌症的治疗[1],其结构示于图1。常用剂型为普通注射液 (Taxol®),其中含有聚氧乙烯蓖麻油溶媒,容易引起过敏性反应,甚至是过敏性休克[2-3]。注射用紫杉醇 (白蛋白结合型) (商品名Abraxane®) 是把紫杉醇和纳米白蛋白颗粒结合在一起的新型纳米制剂[2],用于转移性乳腺癌联合化疗失败后或辅助化疗6个月内复发的乳腺癌的治疗。与普通注射液相比,该制剂不含聚氧乙烯蓖麻油溶媒,具有过敏反应少、静注时间短、作用时间长等优点[4]。紫杉醇在体内主要代谢途径是羟基化,人血浆、胆汁、尿和粪中的主要代谢物是3’-p-羟基紫杉醇和6α-羟基紫杉醇,大鼠肝细胞、肝微粒体和胆汁中的主要代谢物为3’-p-羟基紫杉醇和2-羟基紫杉醇[5-7],在人与大鼠体内都产生3’-p-羟基紫杉醇。3’-p-羟基紫杉醇有一定的抗癌活性和微管稳定作用[8],并保留了原形的一部分骨髓毒性[9],因此对血浆中的紫杉醇及其代谢物3’-p-羟基紫杉醇的药代动力学研究很有意义,可为临床试验提供一定的科学依据。
但以紫杉醇为代表的一些中性化合物因其不含酸性或碱性基团,在质谱中较难离子化,给分析带来了困难。对这些化合物的质谱分析需添加一定的试剂形成加合离子,正离子模式下可用碱金属盐或烷基铵盐[10-13]。Casetta等[11]在测定血清中1α,25(OH)2-维生素D3时,以醋酸锂作为试剂,其加合离子稳定,即使在MS/MS中有H2O损失也不受影响。本工作以碳酸锂作为试剂,利用锂加合离子检测,建立液相色谱-串联质谱(LC-MS/MS)法同时测定大鼠血浆中紫杉醇和其羟基代谢物,并应用于注射用紫杉醇 (白蛋白结合型)的药代动力学研究。
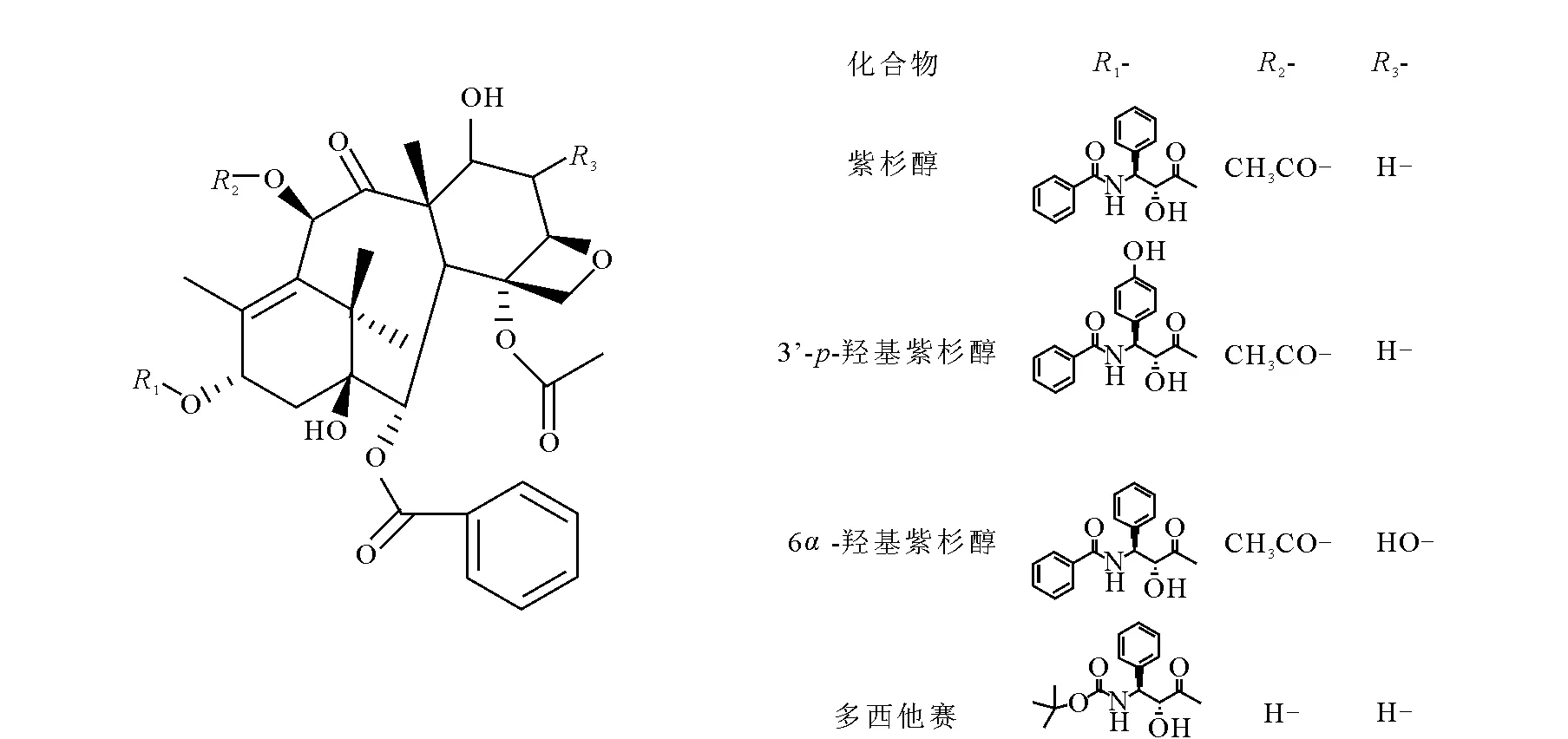
图1 待测物及内标结构式Fig.1 The structures of the analysts and internal standard
1 实验部分
1.1主要仪器与装置
Agilent 6460型三重四极杆串联质谱仪:美国Agilent公司产品,配有电喷雾电离源及Mass Hunter数据采集软件;Agilent 1200液相色谱系统:美国Agilent公司产品;Turbo Vap蒸发仪:美国Zymark公司产品;Milli-Q Gradient超纯水系统:法国Millipore公司产品。
1.2主要材料与试剂
紫杉醇:纯度99.2%,批号11203028,齐鲁制药公司产品;3’-p-羟基紫杉醇:纯度95.0%,批号SLBC4473V,美国Sigma公司产品;多西他赛 (选作紫杉醇测试的内标纯度98.0%,批号100666-201002):中国药品生物制品检定所提供;注射用紫杉醇 (白蛋白结合型):美国Abraxis BioScience公司产品;碳酸锂、甲基叔丁基醚:国药集团化学试剂有限公司产品。甲醇、乙腈(色谱纯):德国Merck公司产品;醋酸铵(色谱纯):德国Fluka公司产品;空白大鼠血浆:上海药物研究所动物实验中心提供。
1.3实验条件
1.3.1色谱条件 色谱柱:Agilent XDB C8柱 (150×4.6 mm×5 μm);C18保护柱 (4.0×3.0 mm×5 μm);流动相:V(乙腈)∶V(10 mmol/L醋酸铵)∶V(100 mmol/L碳酸锂)=60∶40∶ 0.1的混合溶液;流速梯度:0~1.4 min为 1.5 mL/min,1.41~4.2 min为0.65 mL/min,4.21~4.5 min为1.5 mL/min;进样量10 μL;柱温25 ℃。
1.3.2质谱条件 电喷雾离子源 (ESI源),正离子多反应监测 (MRM) 模式,干燥气温度350 ℃,干燥气流速10 L/min,雾化气压力138 kPa,鞘气温度380 ℃,鞘气流速12 L/min,毛细管电压4 500 V,喷嘴电压1 000 V。紫杉醇、3’-p-羟基紫杉醇和内标多西他赛的碰撞能量均为20 V,毛细管出口碰撞电压为200 V。用于定量分析的离子反应分别为m/z860→m/z292 (紫杉醇)、m/z876→m/z(308+515+575) (3’-p-羟基紫杉醇) 和m/z814→m/z288 (内标多西他赛)。
1.4血浆样品预处理
取50.0 μL血浆,分别加入25.0 μL 1.00 mg/L内标多西他赛、100 μL乙腈和100 μL去离子水,涡流混匀,加入3.0 mL甲基叔丁醚,涡流5 min,以3 500 r/min离心5 min 。取上层有机相于另一试管中,40 ℃空气流下吹干,残留物加入100 μL流动相溶解,涡流1 min,进样10 μL。
2 结果与讨论
2.1大鼠血浆中羟基化代谢物的鉴定
采用三重四极杆质谱对大鼠血浆中羟基化代谢物进行定性分析。采用[M+Na]+进行m/z892→m/z892质谱扫描并色谱分离,排除空白基质的影响,再进行二级扫描,得到相应的碎片离子,共检测到2个羟基代谢产物,命名为M1和M2,其色谱图示于图2。其中,M1与3’-p-羟基紫杉醇的质谱碎片相吻合,且色谱行为一致,确定M1为3’-p-羟基紫杉醇;M2的碎片离子为m/z308、547和607,与6α-羟基紫杉醇的质谱碎片相同,但色谱行为不同,因此M2不是6α-羟基紫杉醇。为了排除葡萄糖醛酸结合物源内裂解的干扰,对其进行了质谱监测,结果表明,大鼠血浆中不存在紫杉醇葡萄糖醛酸和硫酸结合物。
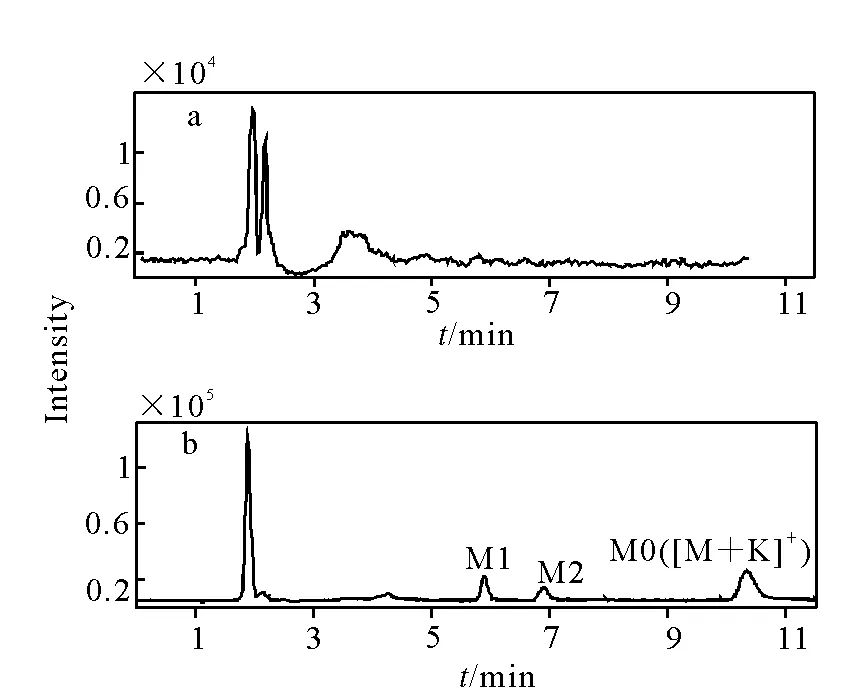
a.空白血浆样品;b.给药后的合并血浆样品图2 大鼠血浆中紫杉醇羟基代谢物色谱图Fig.2 Chromatograms of hydroxylated metabolites of paclitaxel in rat plasma
2.2质谱条件优化
将紫杉醇用乙腈稀释至一定浓度,在不同流动相下进行一级全扫描,其质谱图示于图3。在未添加碱金属离子的流动相中,(V(乙腈)∶V(10 mmol/L醋酸铵)=60∶40的混合溶液为原始流动相)基峰离子为[M+Na]+,其余加合离子为[M+H]+和[M+K]+,[M+H]+的质谱响应较小。添加Li2CO3至0.1 mmol/L后,[M+Li]+为主要加合离子,质谱响应显著增加。紫杉醇是一个中性化合物,对比未添加碱金属离子流动相中[M+H]+和[M+Na]+两天的质谱响应,[M+Na]+质谱响应增加1倍,而[M+H]+响应几乎不变。加入不同量的甲酸观察其对[M+Na]+响应的抑制,抑制效果均不佳,其羟基代谢物结果类似。
对紫杉醇不同加合离子进行二级产物离子扫描,其质谱图示于图4。[M+Li]+、[M+Na]+和[M+K]+主要碎片离子分别比[M+H]+的碎片离子大6、22和38 u,推测断裂方式均为C13位侧链酯键断裂,裂解的碎片离子均加合碱金属离子。不同加合离子的碰撞能量列于表1,由表1可知,[M+Li]+的碰撞能量均低于[M+Na]+和[M+K]+。在低碰撞能量下(5 V),[M+Li]+产生少量碎片离子,而[M+Na]+几乎不能打碎;在高碰撞能量下(50 V),[M+Li]+的碎片信息较[M+Na]+丰富,适用于较高分子质量的中性化合物结构分析与测定。3’-p-羟基紫杉醇和内标的二级全扫描质谱图示于图5。
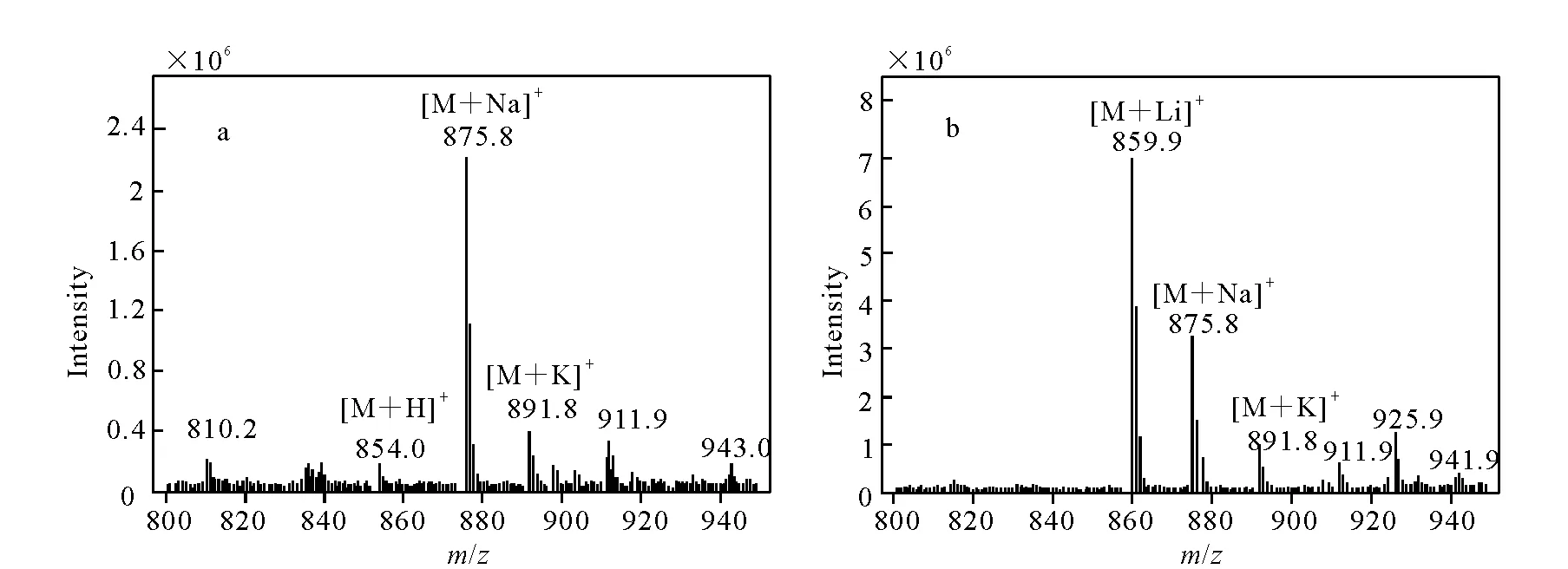
a.未添加碱金属离子;b.添加碳酸锂至0.1 mmol/L图3 紫杉醇在不同流动相下的一级全扫描质谱图Fig.3 Full-scan mass spectra of paclitaxel in different mobile phases
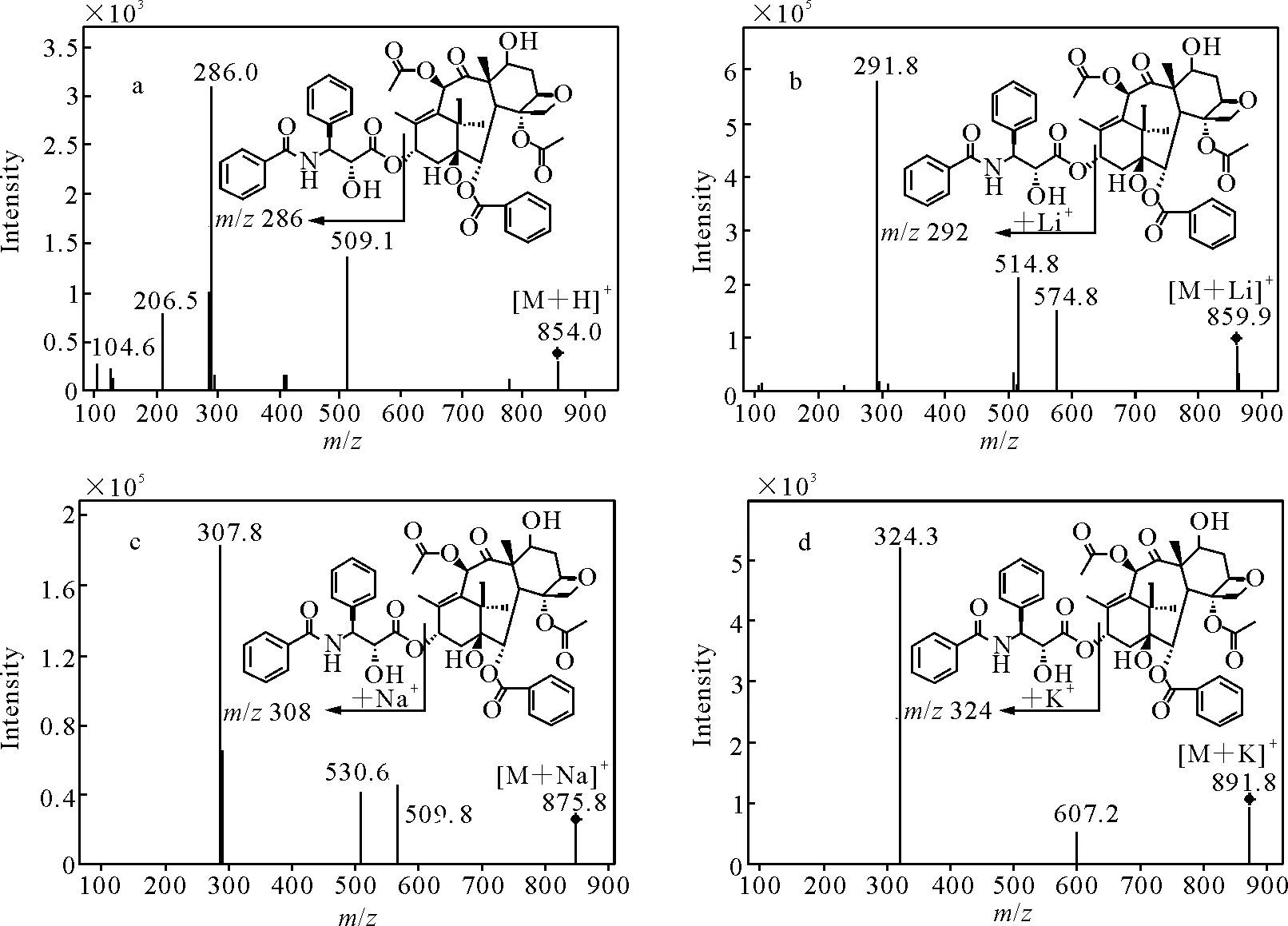
a.[M+H]+; b.[M+Li]+; c.[M+Na]+; d.[M+K]+图4 紫杉醇不同加合离子的二级全扫描质谱图Fig.4 Product ion mass spectra of different adduct ions for paclitaxel
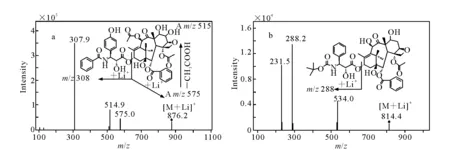
图5 3’-p-羟基紫杉醇(a)和内标多西他赛(b)的[M+Li]+二级全扫描质谱图Fig.5 Product ion mass spectra of [M+Li]+ for 3’-p-hydroxypaclitaxel (a) and internal standard docetaxel (b)

表1 紫杉醇不同加合离子的碰撞能量
2.3色谱条件优化
紫杉醇溶液在代谢物通道m/z876→m/z308产生与原形保留时间一致的色谱峰,对分析造成一定的干扰。可能的原因为[M+Li]+、[M+Na]+、[M+K]+质荷比相差16 u,而3’-p-羟基紫杉醇与原形质荷比也相差16 u,详细数据列于表2。由于其产物离子分别加合了Li+、Na+、K+,因此质谱无法区分,待测物之间需要进行完全的色谱分离。通过降低有机相比例,延长保留时间,可以将其完全分离,总分析时间为4.5 min。人体中的主要羟基化代谢物为6α-羟基紫杉醇和3’-p-羟基紫杉醇,在该色谱条件下,两者的色谱行为较接近,但其碎片离子不同,所以此色谱方法也适用于人血浆中紫杉醇和羟基代谢物的同时测定。
有文献[14]报道采用0.5 mmol/L碳酸锂作为流动相添加剂测定人血浆中怕伐他汀,考虑到碳酸锂为非挥发性盐类,且在乙腈中的溶解度较低,本实验将碳酸锂的用量降至0.1 mmol/L。
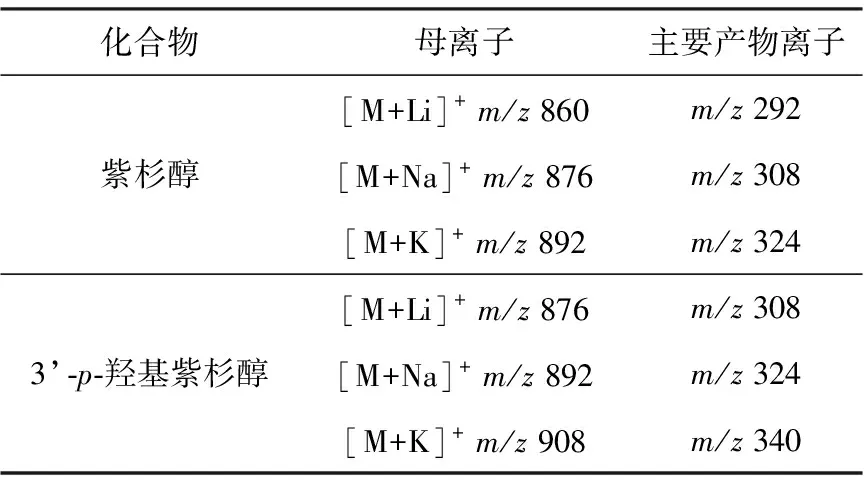
表2 紫杉醇及3’-p-羟基紫杉醇的主要产物离子
2.4预处理方法的选择
文献在测定血浆中的紫杉醇及其羟基代谢物时采用液-液萃取或固相萃取进行血浆预处理,在方法建立初期曾采用沉淀蛋白法,但基质效应较强[15-20]。为避免此现象,采用液-液萃取,并对提取试剂进行考察。血浆样品用V(甲基叔丁醚)∶V(乙醚-二氯甲烷)=2∶1 提取后进行LC-MS/MS分析,甲基叔丁醚的提取效率较高且稳定,选用为提取试剂。
2.5方法学确证
2.5.1方法的专属性 分别取6只大鼠的50.0 μL空白血浆,除不加内标溶液外,按1.4项操作,得色谱图6a;将一定浓度的标准溶液和内标溶液加入空白血浆中,依同法操作,得色谱图6b;取大鼠约药后的50.0 μL血浆样品,按1.4项操作,得色谱图6c。结果表明,空白血浆中的内源性物质不干扰待测物及内标的测定。
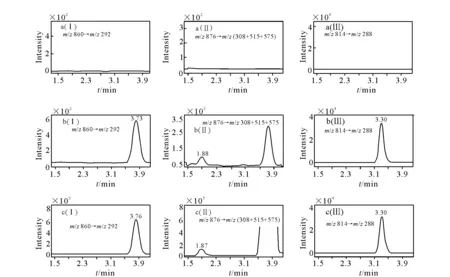
a.空白血浆样品;b.空白血浆中加入紫杉醇至1.00 μg/L、3’-p-羟基紫杉醇至0.50 μg/L、内标多西他赛至500 μg/L;c.1号大鼠静脉注射15 mg/kg注射用紫杉醇 (白蛋白结合型) 15 min后的血浆样品图6 LC-MS/MS法测定紫杉醇(I)、3’-p-羟基紫杉醇(II)和内标多西他赛(III)的典型MRM色谱图Fig.6 Chromatograms of paclitaxel (I), 3’-p-hydroxypaclitaxel (II) and internal standard (III)
2.5.2线性范围和定量下限 取50.0 μL空白血浆,分别加入50.0 μL紫杉醇和3’-p-羟基紫杉醇标准系列溶液(紫杉醇/3’-p-羟基紫杉醇相对于血浆药物浓度为1.00/0.50,3.00/1.00,10.0/2.50,30.0/5.00,100/15.0,300/50.0,1 000,3 000,8 000 μg/L),按1.4方法处理后进样分析,建立标准曲线。以血浆中待测物浓度x(μg/L)为横坐标,待测物与内标的峰面积y为纵坐标,用加权最小二乘法 (W=1/x2) 进行回归计算,得回归方程,即标准曲线。紫杉醇典型的回归方程为y=6.49×10-3x+7.00×10-3,R2=0.990 5;3’-p-羟基紫杉醇典型的回归方程为y=4.28×10-3x-4.45×10-4,R2=0.994 7。
取50.0 μL定量下限样品,按1.4项操作,连续3天进行6样本分析,紫杉醇和3’-p-羟基紫杉醇的日内、日间精密度 (RSD)均小于11.3%,准确度 (RE) 为-3.8%~0.6%,该方法的紫杉醇和3’-p-羟基紫杉醇的定量下限分别可达1.00和0.50 μg/L。
2.5.3精密度和准确度 取50.0 μL QC样品(紫杉醇/3’-p-羟基紫杉醇血浆浓度分别为10.0/1.00,100/15.0和6 400/40 μg/L),按1.4项操作,每一浓度进行6样本分析,连续测定3天。紫杉醇和3’-p-羟基紫杉醇的QC样品的日内、日间精密度均小于14.3%,准确度在-4.5%~2.3%之间。
2.5.4提取回收率和基质效应考察 待测物及内标的提取回收率均在92.5%~107%之间,RSD均小于13.9%;基质效应均在86.2%~104%,RSD均小于13.5%。表明本实验的LC-MS/MS条件可有效地避免大鼠血浆基质效应。
2.5.5稳定性考察 考察低、高浓度QC样品室温放置6 h、血浆样品处理后室温放置24 h及3次冻融稳定性 (-70 ℃至室温),每一浓度水平进行3样本分析。在上述条件下,紫杉醇和3’-p-羟基紫杉醇的准确度均在-9.2%~8.8%之间,精密度小于11.2%。
2.5.6稀释试验 取稀释QC样品(紫杉醇血浆浓度为32.0 mg/L),用大鼠空白血浆稀释5倍,进行6样本分析。紫杉醇的精密度为9.4%,准确度为6.7%。结果表明,血浆样品经空白血浆稀释5倍后,不影响测定结果的准确性。
2.6方法应用
取3只成年、健康的雄性SD大鼠,体重180~220 g,年龄7~8周,静脉推注给予15 mg/kg注射用紫杉醇 (白蛋白结合型)。采集给药前及给药后2 min、5 min、15 min、0.5 h、1.0 h、2.0 h、4.0 h、6.0 h、10 h、24 h和48 h血样,分离血浆置于-70 ℃保存。原形紫杉醇和3’-p-羟基紫杉醇的平均血浆浓度-时间曲线示于图7。实验数据经WinNonlin5.3软件处理,采用非房室计算主要药动学参数,结果列于表3。
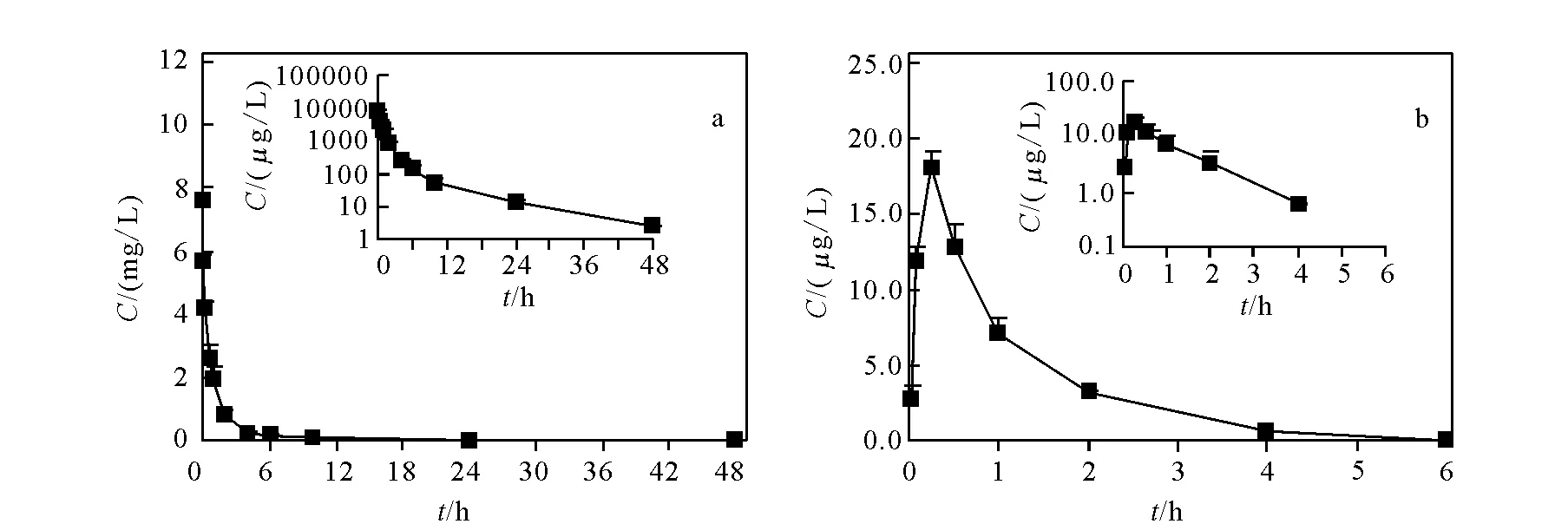
图7 受试大鼠推注给予注射用紫杉醇 (白蛋白结合型) 后,紫杉醇(a)和3’-p-羟基紫杉醇(b)的平均血浆浓度-时间曲线Fig.7 Mean plasma concentration versus time curves for paclitaxel (a) and 3’-p-hydroxypaclitaxel (b) in rats
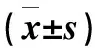
表3 紫杉醇及3’-p-羟基紫杉醇的 主要药动学参数
3 结论
本研究建立了以碳酸锂作为试剂,同时测定大鼠血浆中紫杉醇和羟基代谢物的LC-MS/MS方法。锂加合离子稳定性好、碰撞能量低、灵敏度高,适用于一些中性化合物的定量和结构分析。本方法灵敏度高,血浆用量为50.0 μL,紫杉醇定量下限为1.00 μg/L,3’-p-羟基紫杉醇定量下限为0.50 μg/L,能够满足紫杉醇及其羟基代谢物的药动学研究。
[1] FERLINI C, OJIMA I, DISTEFANO M, et al. Second generation taxanes: From the natural framework to the challenge of drug resistance[J]. Curr Med Chem Anti-Cancer Agents, 2003, 3: 133-138.
[2] MIELE E, SPPINELLI G P, MIELE E, et al. Albumin-bound formulation of paclitaxel (Abraxane®ABI-007) in the treatment of breast cancer [J]. Int J Nanomedicine, 2009, (4): 99-105.
[3] GARDERN E R, DAHUT W L, SCRIPTURE C D. Randomized crossover pharmacokinetic study of solvent-based paclitaxel and nab-paclitaxel [J]. Clin Cancer Res, 2008, 14(13): 4 199-4 205.
[4] GRADISHAR W J. Albumin-bound paclitaxel: A next-generation taxane[J]. Expert Opin Pharmacother, 2006, 7(8): 1 041-1 053.
[5] JAMIS-DOW C A, KLECKER R W, KATKI AG,et al. Metabolism of taxol by human and rat liver in vitro: a screen for drug interactions and interspecies differences[J]. Cancer Chemother Pharmacol, 1995, 36: 107-114.
[6] ANDERSON C D, WANG J, KUMAR G N, et al.Dexamethason induction of taxol metabolism in the rat[J]. Drug Metab Dispos, 1995, 23: 1 286-1 290.
[7] MONSARRAT B, MARIEL E, Cros S, et al. T-axol metabolism isolation and identification of three major metabolites of taxol in rat bile[J]. Drug Metab Dispos, 1990, 18(6): 895-901.
[8] ARORA V. Antisense strategies for redirection of drug metabolism:Using paclitaxel as a model[J]. Methods Mol Med, 2004, 106: 273-292.
[9] SPRATLIN J, SAWYER M B. Pharmacogenetics of paclitaxel metabolism[J]. Crit Rev Oncol/Hematol, 2007, 61: 222-229.
[10] CECH N B, ENKE C G. Practical implications of some recent studies in electrospray ionization fundamentals[J]. Mass Spectrom Rev, 2001, 20: 362-387.
[11] CASETTA B, JANS I, BILLEN J, et al. Development of a method for the quantification of 1α,25(OH)2-vitamin D3in serum by liquid chromatography tandem mass spectrometry without derivatization[J]. Eur J Mass Spectrom, 2010, 16(1): 81-89.
[12] TARVAINEN M, PHUPHUSIT A, SUOMELA J P, et al. Effects of antioxidants on rapeseed oil oxidation in an artificial digestion model analyzed by UHPLC-ESI-MS[J]. J Agric Food Chem, 2012, 60: 3 564-3 579.
[13] HAMILTON J F, LEWIS A C, CAREY TJ, et al.Characterization of polar compounds and oligomers in secondary organic aerosol using liquid chromatography coupled to mass spectrometry[J]. Anal Chem, 2008, 80: 474-480.
[14] BECQUEMONT L, FUNCK-BRENTANO C,JAILLO P. Mibefradil, a potent CYP3A inhibitor, does not alter pravastatin pharmacokinetics[J]. Fundam Clin Pharmacol, 1999, 13: 232-236.
[15] ALEXANDER M S, KISER M M, CULLEY T, et al. Measurement of paclitaxel in biological matrices: High-throughput liquid chromatographic-tandem mass spectrometric quantification of paclitaxel and metabolites in human and dog plasma[J]. J Chromatogr B, 2003, 785: 253-261.
[16] ZHANG W, DUTSCHMAN G E, LI X, et al. Quantitation of paclitaxel and its two major metabolites using a liquid chromatography-electrospray ionization tandem mass spectrometry[J]. J Chromatogr B, 2011, 879: 2 018-2 022.
[17] VAINCHTEIN L D, THIJSSEN B, STOKVIS E, et al. A simple and sensitive assay for the quantitative analysis of paclitaxel and metabolites in human plasma using liquid chromatography/tandem mass spectrometry[J]. Biomed Chromatogr, 2006, 20: 139-148.
[18] HENRIK G, KARIN V, BJORN N, et al. Mea-surement of paclitaxel and its metabolites in human plasma using liquid chromatography/ion trap mass spectrometry with a sonic spray ionization interface[J]. Rapid Commun Mass Spectrom, 2006, 20: 2 183-2 189.
[19] YAMAGUCHI H, FUJIKA W A, ITO H, et al.Quantitative determination of paclitaxel and its metabolites, 6α-hydroxypaclitaxel and p-3'-hydroxypaclitaxel, in human plasma using column-switching liquid chromatography/tandem mass spectrometry[J]. Biomed Chromatogr, 2012, 27(4): 539-544.
[20] MORTIER K A, RENARD V, VERSTRAETE A G, et al. Development and validation of a liquid chromatography-tandem mass spectrometry assay for the quantification of docetaxel and paclitaxel in human plasma and oral fluid [J]. Anal Chem, 2005, 77: 4 677-4 683.
DeterminationofPaclitaxelandHydroxylatedMetabolitesinRatPlasmawithLithiumAdductIonbyLC-MS/MS
FAN Ya-xin1,2, CHEN Xiao-yan2, MA Zhi-yu2, GAO Zhi-wei2, ZHONG Da-fang1,2
(1.ZhejiangUniversityofTechnology,Hangzhou310014,China;2.ShanghaiInstituteofMateriaMedica,ChineseAcademyofSciences,Shanghai201203,China)
A liquid chromatography-tandem mass spectrometry (LC-MS/MS) method was explored for improving the sensitivity of paclitaxel and 3’-p-hydroxypaclitaxel in rat plasma with metal ion. Paclitaxel is easy to produce alkali metal adduct ions (Li+, Na+, K+) during mass spectrometry analysis. The mass spectrometry response of [M+H]+is low and the stability of [M+Na]+is poor without adding metal ions in the mobile phase. In this study, paclitaxel and hydroxylated metabolites were quantative analyzed using lithium carbonate reagent. The linear concentration ranges of the calibration curves for paclitaxel and 3’-p-hydroxypaclitaxel are 1.00—8 000 μg/L and 0.50—50 μg/L, respectively. The method is sensitive, simple and rapid, which is suitable for the pharmacokinetic study of the albumin-bound (nab-)paclitaxel. Lithium adduct ion is stable, high mass spectrometry response and low collision energy, is suitable for the quantative and structural analysis of some neutral compounds.
lithium adduct ion; paclitaxel; hydroxylated metabolites; liquid chromatography-tandem mass spectrometry
O 657.73
A
1004-2997(2013)05-0137-08
10.7538/zpxb.2013.34.03.0137
2012-12-11;
2013-03-13
范亚新,女,浙江人,硕士研究生,从事药物代谢与药物动力学研究。E-mail: fanyaxin20080908@126.com
钟大放,男,辽宁人,研究员,从事药物代谢与药物动力学研究。E-mail: dfzhong@mail.shcnc.ac.cn
猜你喜欢
现代临床医学(2022年4期)2022-09-29 07:36:10
口腔护理用品工业(2021年4期)2021-11-02 08:22:54
中国特种设备安全(2021年12期)2021-04-26 14:37:00
中成药(2018年6期)2018-07-11 03:01:32
实用口腔医学杂志(2017年6期)2017-09-19 02:51:06
中外医疗(2016年15期)2016-12-01 04:25:50
中国粮油学报(2016年5期)2016-01-23 02:45:06
分析测试学报(2015年7期)2016-01-13 06:19:16
哈尔滨医药(2015年2期)2015-12-01 03:57:41
质谱学报(2015年5期)2015-03-01 03:18:37
