
CO32-/Co2+/Ni2+浓度对混合相纳米Ni(OH)2物化性能的影响
2013-10-17 03:03庄义环朱燕娟赵汝冬林晓然刘泳林张树杰许庆胜
无机化学学报 2013年2期
庄义环 朱燕娟 赵汝冬 林晓然 刘泳林 张树杰 许庆胜
(广东工业大学物理与光电工程学院,广州 510006)
0 引 言
镍氢电池容量由正极决定,因此研究正极活性材料氢氧化镍(Ni(OH)2)性能,提高电池容量一直受到人们的关注。研究表明[1-3],制备条件对氢氧化镍的物化性能产生重要影响,通过优化条件可以有效提高其电化学性能。氢氧化镍有α和β两种晶型,与β-Ni(OH)2相比,α-Ni(OH)2具有更高放电容量和无机械形变等优异特性[4],但α-Ni(OH)2在强碱中稳定性较差且容易转变成β-Ni(OH)2。为了解决这一问题,国内外学者做了大量工作,常用的方法是通过掺杂金属离子或稀土元素,如用 Al[5-6]、Co[7-8]、Zn[9]、Fe[10]、Y[11]或多种元素复合取代Ni离子[3,12]以达到稳定晶格的目的。
上述研究主要集中在掺杂元素、掺杂比例及镍源的优化上,对稳定反应液pH值的研究少有报道。由于氨水的弱碱性使其对pH值调节作用偏弱,且易挥发使反应后期失去调控作用。溶液的pH值直接影响材料的形貌、粒径及其性能,因此,保持稳定的pH值至关重要。本研究用无水碳酸钠(Na2CO3)作为缓冲剂,与氨水协同作用使溶液pH值保持稳定,同时通过调节Na2CO3用量可以有效改变氢氧化镍晶型结构。
本研究主要内容:采用自主知识产权的超声波辅助沉淀法[13]制备Co掺杂纳米混合相Ni(OH)2,研究了Co掺杂比例、缓冲剂Na2CO3用量和反应物Ni2+浓度对Ni(OH)2的晶相结构、形貌、粒径及电化学性能的影响。鉴于成本考虑,本研究只用8wt%的纳米样品掺入到工业用微米级β球镍中,研究其复合电极的电化学性能,并探讨了复合电极充放电机理。
1 实 验
改变 Na2CO3用量为 0.5 g(0.047 mol·L-1),其它原料用量、溶液体积及制备条件与上述C样品相同,制得的样品标为G。
改变溶液体积为50 mL,Na2CO3用量分别取0.4、0.5、0.6 g(浓度依次为 0.075、0.094、0.113 mol·L-1),其它原料用量及制备条件与上述C样品相同,制得的样品标为 D、E、F。 据此,样品 D、E、F的 Ni2+浓度为 0.4 mol·L-1,而样品 G 的 Ni2+浓度为 0.2 mol·L-1,4 个样品的 nCo2+/nNi2+比均为25%。虽然样品E和G的Na2CO3用量和Co2+掺杂量相同,但因溶液体积增大1倍,使得样品G的Ni2+、Co2+及Na2CO3物质的量浓度均比样品E低1倍。
分别取上述样品D、E、F、G与工业用微米级球镍、镍粉、PTFE(聚四氟乙烯)、CMC(羧甲基纤维素)按质量分数 8%∶86%∶3%∶2%∶1%的比例配料,加入适量蒸馏水搅拌混合成糊状物负载到泡沫镍基片中,然后干燥压片,制成复合镍正极依次标为d、e、f、g。负极用储氢合金粉、CMC、镍粉按质量分数比为88%∶2%∶10%混合制作而成,制作方法与正极相同。电极d~g含Co质量分数均为1.02wt%。为了对比,用同样的方法制作了纯β球镍电极h。
将制成的正极、负极与Hg/HgO参比电极组装成三电极体系,用上海辰华仪器CHI 660C电化学工作站在室温下进行伏安特性测试,测试电压范围为-0.20.7 V。将正极片置于二负极片之间制成模拟电池,采用新威BTS-51800型电池性能测试仪进行充放电性能测试,充放电制度取0.5C,放电电压均截至 1.0 V。
表征样品结构、粒径及形貌的仪器分别为:日本理学D/max-ⅢA全自动X射线衍射仪(以Cu靶Kα为辐射源,波长为 0.154 06 nm,工作电压 36 kV,电流 20 mA,扫描速度 2°·s-1)、激光粒度仪(美国Microtrac,Nanotrac 150)、荷兰 Philips Tecnai 10 透射电子显微镜。
2 结果与讨论
2.1 掺Co比例对样品晶相结构的影响
图1是不同掺Co比例样品A、B、C的XRD图。图中可见,各样品在衍射角11°和38°出现了衍射峰,与 α-Ni(OH)2标准图谱 PDF No.38-0715(003)和(015)晶面特征峰一致;在衍射角 33°、38°、52°、59°及62°出现的衍射峰与 β-Ni(OH)2标准图谱 PDF No.14-0117 一致; 图中衍射角 20.5°的峰是 β-Ni(OH)2在(001)晶面的 19°特征峰和 α-Ni(OH)2在(006)晶面的23°特征峰的叠加结果,而38°衍射峰是由β相(101)特征峰和α相(015)特征峰叠加而成。由此可知,3个样品均为α相和β相混合的氢氧化镍。
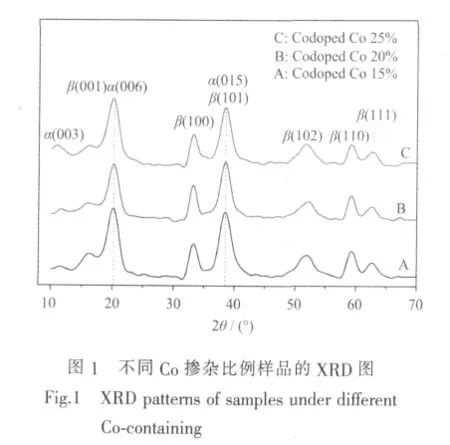
图1显示,随Co掺杂比例的提高,11°的3个衍射峰相对强度增强,38°处衍射峰强度减弱。衍射角11°和38°的峰分别是α相和β相的主要特征峰,其强度变化反映了两种晶相氢氧化镍含量的变化,这意味着随着Co含量的增加,α-Ni(OH)2所占比例增大。C样品在11°的衍射峰较强,因此,掺Co物质的量比为25%的样品其α-Ni(OH)2比例较大。
2.2 Na2CO3对晶相结构、形貌、粒径及电化学性能的影响
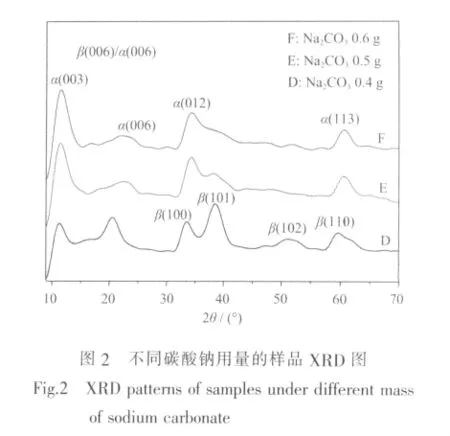
为了提高样品α-Ni(OH)2比例,本研究通过改变Na2CO3用量的方法进行试验。结果表明,当Na2CO3用量由 0.08 g(A、B、C 样品) 增加到 0.4 g(D样品)时,11°衍射峰明显增强,继续增加Na2CO3用量,作为 α-Ni(OH)2特征的(003)、(006)、(012)、(113)晶面的衍射峰不断增强(如图2),而标示β-Ni(OH)2特征的 (100)、(101)、(102)、(110) 晶面衍射峰却不断减弱,衍射角20.5°的叠加峰逐渐转化为(006)晶面的α-Ni(OH)2特征峰,意味着 19°(001)晶面的 β-Ni(OH)2特征峰逐渐消失。当Na2CO3用量为0.6 g时 (F样品),(101)晶面衍射峰已难以分辨,而出现 34°~38°范围的不对称衍射峰,这是α-Ni(OH)2螺旋层状结构的特征峰[14]。 由此可见,样品 D 和 E(0.5 g Na2CO3)是α相和β相混合的氢氧化镍,而样品F是典型的α-Ni(OH)2,样品E的α-Ni(OH)2比例或相丰度大于样品D。
透射电镜测试表明,样品D、E、F、G的微观形貌均为针状(图3)。图3可见,随着Na2CO3用量增加,一次颗粒长径比变小,长度从约200 nm减小到60~70 nm,径向在10~40 nm之间。一次颗粒长径比的减小,其颗粒的比表面积增大,活性提高,团聚加重,因此,二次颗粒的粒径从D~F依次增大。用激光粒度仪测得的D、E、F样品粒度分布如图4所示,其平均粒径分别为79、63、75 nm。由于激光粒度仪测得的粒径是一次和二次颗粒的平均粒径,使得样品E的平均粒径最小。

在反应液中加入Na2CO3有两个作用,一是提供阴离子 CO32-、HCO3-(如(1)、(2)式),同时对反应液 pH值起到缓冲稳定作用,故称其为缓冲剂。当溶液pH值降低时,反应(2)向右进行,反之,(2)式向左进行,从而保持反应液pH值的稳定。
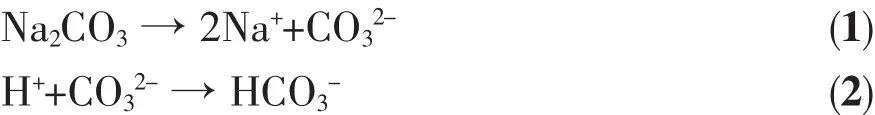
在制备氢氧化镍过程中掺入Co元素后,Co2+部分取代Ni2+或间隙式嵌入Ni(OH)2四面体间隙,使其晶胞正电荷过剩,为保持电中性,晶体层间将吸入阴离子导致层间距增大,从而形成α-Ni(OH)2晶相。被吸入晶体层间的阴离子除了Cl-、NO3-之外,根据反应式(1)、(2),还有 CO32-和 HCO3-。 随着 Na2CO3用量的增加,CO32-和HCO3-浓度增大。根据第2节实验条件,D、E、F 三样品的 NiCl2和 Co(NO3)2用量相等,溶液的 Cl-、NO3-数量相同,在这种情况下,CO32-和HCO3-增加,给晶体层间提供的阴离子数增多,从而有利于 α-Ni(OH)2形成,导致样品 D、E、F的 α-Ni(OH)2相丰度依次增大。体系的Ni(OH)2成核速度随着Na2CO3用量增加而增大,使成核速度大于晶粒生长速度,这样又使得样品D、E、F晶粒长径比依次减小(图 3)。
图5是含有8wt%纳米样品的复合电极d、e、f和纯β球镍电极h在0.5C倍率下的充放电曲线,其放电比容量分别为 285.7、306.9、290.6、235.3 mAh·g-1。可见,随着Na2CO3用量的增加,放电比容量先增大后减小,当用量为0.5 g时,电极e的放电比容量最大。放电比容量与两个因素密切相关,一是α-Ni(OH)2的相丰度,二是团聚程度及粒径,研究显示[15],放电比容量随粒径减小而增大。电极d的放电比容量低于电极e,显然是由于电极d中的样品D所含的α-Ni(OH)2比例较低且粒径较大所致。对于电极f,虽然其所含的纳米样品F几乎为纯α-Ni(OH)2,但放电比容量却不如电极e,主要原因是样品F晶粒间团聚较重(图3),活性降低,同时样品F较大的平均粒径(图4)使其与β球镍混合时没有像样品E与球镍混合时那么匹配,不能较充分地填充到球镍的空隙中,因此颗粒间的接触电阻相对较大,从而降低了电极活性材料的利用率,导致电极f放电比容量降低。图5还表明,缓冲剂Na2CO3对电极充放电电位也产生一定影响,其用量增加,充电平台降低,放电平台稍升高。
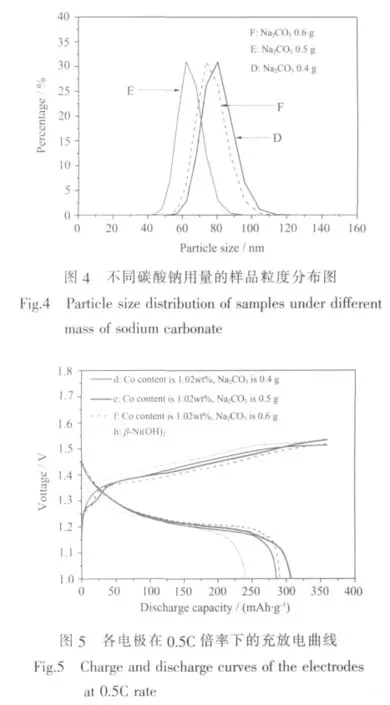
Nethravathi等[7]报道,将 Co 取代 α-Ni(OH)2样品以60wt%比例与添加剂混合制成电极时,其放电比容量达到 373 mAh·g-1;文献[8]报道,含有 90wt%比例Co取代α-Ni(OH)2纳米样品的电极,其放电比容量最高达 415 mAh·g-1。本研究的复合电极 e只含8wt%的纳米样品,放电比容量达到306.9 mAh·g-1,比纯球镍电极h高71.6 mAh·g-1,折算成纯纳米样品电极的克容量将达到895 mAh·g-1,比α-Ni(OH)2的理论容量(482 mAh·g-1)高 413 mAh·g-1,对于该结果的解释主要有以下两个方面。
一是纳米颗粒掺入到微米级球镍中,填充了微米β-Ni(OH)2颗粒间的空隙,使颗粒间接触电阻减小,缩短了质子在固相中的扩散距离。根据电极反应的固相质子扩散机理[16,17],质子扩散路径的缩短,减小了镍电极极化,提高了充电效率和活性物质的利用率。而电极中纳米样品的Co既提高了电极的导电性,又改善了其充放电过程的可逆性[18],使上述作用得到放大,从而有效提高复合电极的放电比容量。
二是由于纳米样品具有很强的催化活性,在电极反应过程中,掺入球镍中的纳米α-Ni(OH)2同时充当活化中心,促使其邻近的β-Ni(OH)2吸附阴离子转化为α-Ni(OH)2,其反应式如下:
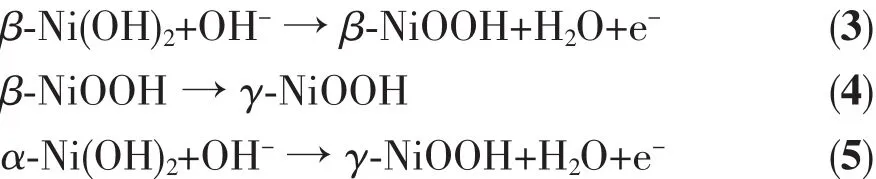
其中反应(3)和(5)是可逆过程,(4)是不可逆过程。由于复合电极中球镍β-Ni(OH)2占86wt%,纳米样品占8wt%(α-Ni(OH)2含量不大于8wt%),根据反应动力学,放电过程中其(3)、(4)反应很容易发生,产生了大量的γ-NiOOH,在纳米α-Ni(OH)2催化作用下,使反应(5)的逆反应可以充分进行。因此,在充放电过程中,复合电极的α-Ni(OH)2远大于8wt%,使放电比容量得到较大提高。当然,对纯β-Ni(OH)2电极h,上述反应也会发生,但由于本身没有α-Ni(OH)2作为活化中心,使得由γ-NiOOH转化来的α-Ni(OH)2数量少而且极不稳定,很容易脱出阴离子还原为β-Ni(OH)2,同时由于纯β-Ni(OH)2电极充放电过程是在β-Ni(OH)2与β-NiOOH之间电对循环,过冲时很容易被转化成体积较大的γ-NiOOH,导致不可逆的电极膨胀,使电极性能衰退[19-20],因此纯球镍电极h放电比容量低。
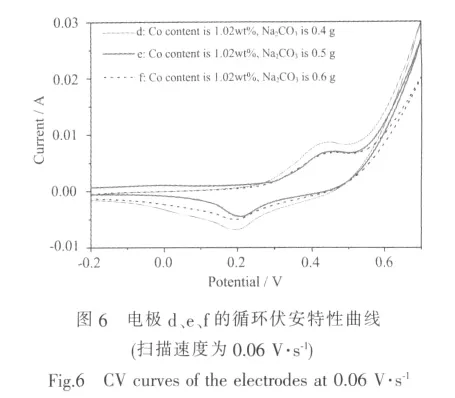
图 6 是复合电极 d、e、f在 0.06 V·s-1的扫描速度下测得的循环伏安特性曲线,表1列出了各峰电压值。由图表可以看出,随着Na2CO3用量的增加,三电极的氧化峰和还原峰电压之差(EO-ER)先减小后增大,电极e的差值最小,为0.258V。EO-ER代表了电极反应的可逆性[21],差值越小,电化学反应可逆性越好,电极极化越小,因此电极e的可逆性最好,在电极反应过程中将有更多的活性物质被利用。析氧峰和氧化峰电压之差EOER-EO随着Na2CO3用量的增加先增大后减小。EOER-EO的差值体现电极析氧难易程度,差值越大表示充电效率越高[5]。电极e的EOER-EO比电极d高11 mV,但电极e和f的EOER-EO差值几乎相等 (这与图5二电极充电曲线几乎重合相符),表明电极e和f的充电效率较高,它们比电极d不容易析氧。由此可见,添加适量的Na2CO3既有利于提高电极的可逆性,又有助于抑制析氧反应的发生。
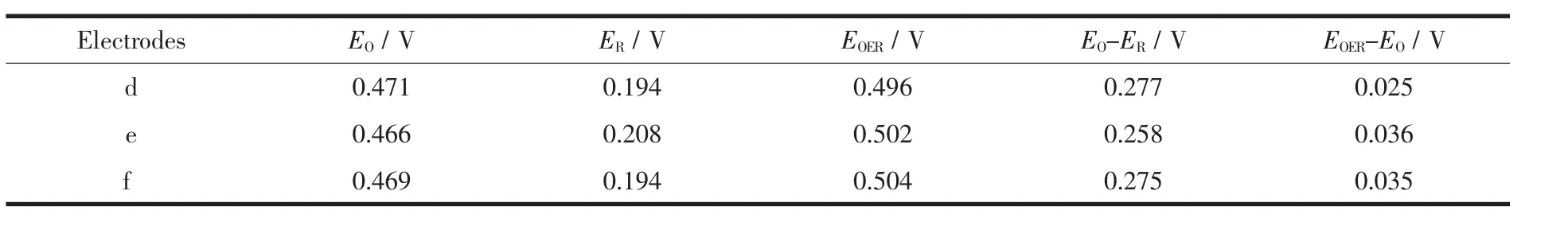
表1 电极的循环伏安特性参数(扫描速度为0.06 V·s-1)Table 1 CV date of the electrodes tested at 0.06 V·s-1
2.3 反应物浓度对样品物化性能的影响
除了缓冲剂,本文还对比了反应物浓度对样品结构、形貌、粒径及充放电性能的影响。样品E、G(制备条件如“1实验”)的微观形貌如图3E、3G,其XRD图和粒度分布如图7、8所示,图9是相应电极e、g的充放电曲线。
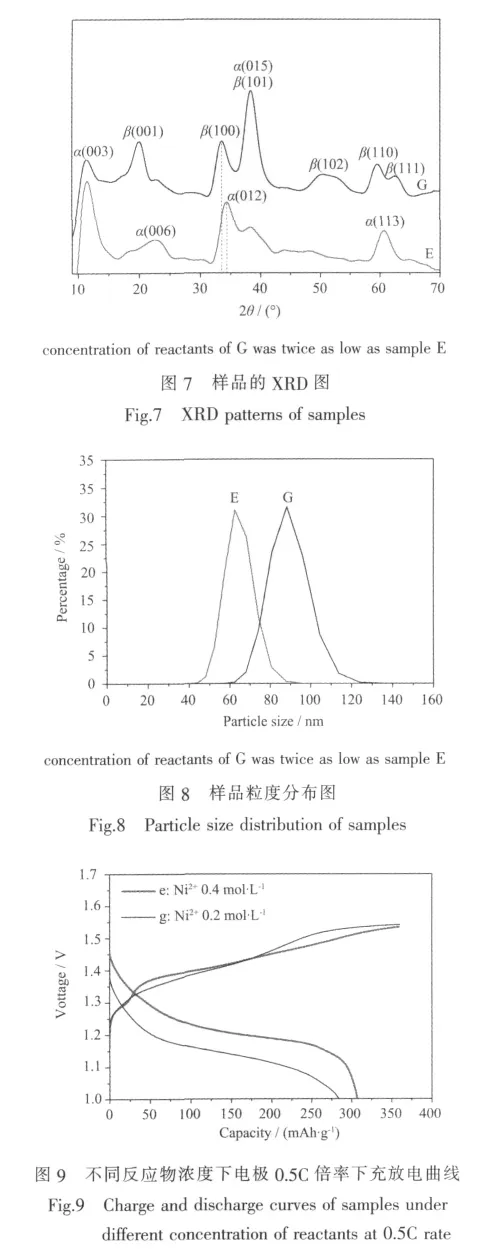
从图7可见,样品G在11°出现的 α-Ni(OH)2特征峰峰强较弱,而β相的特征峰全部出现且峰强较强,所以样品G也是和的混合相结构,但以β-Ni(OH)2为主,而样品E则是以α-Ni(OH)2为主。由此可知,较高的反应物浓度有助于α-Ni(OH)2生成。样品G与E的形貌也存在明显差别,但G与D的形貌相仿(如图3)。样品G的径向略大于样品D,其平均粒径为88 nm,大于样品D粒径(79 nm,图2D)和样品E粒径(63 nm,图8E)。此外,样品G与D的XRD图类似(图7G和图2D),表明反应物浓度Ni2+、掺杂Co2+浓度及缓冲剂Na2CO3浓度较低时均易生成β-Ni(OH)2。
图9显示,含样品E、G的电极e、g在0.5C倍率下的放电比容量分别为306.9 mAh·g-1和283.9 mAh·g-1,且前者放电平台远高于后者。显然,这也是由于电极e含α-Ni(OH)2的相丰度较电极g高且前者粒径小于后者导致的结果。
3 结 论
(1)用超声波辅助沉淀法制备的Co掺杂纳米氢氧化镍具有α和β相混合结构。混合相中α-Ni(OH)2相丰度随Co掺杂比例增大而增大,当Co掺杂物质的量比为25%时,其α-Ni(OH)2所占比例较大。
(2)缓冲剂Na2CO3用量对生成物的晶相结构、形貌、粒径及电化学性能均产生重要影响。Na2CO3用量增加,混合相中α-Ni(OH)2所占比例增大,针状的晶粒长径比减小。当Na2CO3用量增加到一定程度,样品结构趋向纯α-Ni(OH)2,但颗粒的团聚程度加重。添加适量的Na2CO3才能使材料的电化学性能达到最佳。当Na2CO3用量为0.5 g时,其复合电极(Co含量1.02wt%)的充电效率最高,放电比容量最大,达到 306.9 mAh·g-1。
(3)较高的反应物浓度有利于α-Ni(OH)2生成,使其电化学性能得到更好的体现。
[1]Jayashree R S,Kamath P V.J.Power Sources,2002,107(1):120-124
[2]Han X J,Xie X M,Xu C Q,et al.Opt.Mater.,2003,23(1/2):465-470
[3]Liu C J,Wu H B,Li Y W.Phys.Chem.Solid,2009,70(3/4):723-726
[4]Jayashree R S,Vishnu Kamath P.J.Power Sources,2002,107(1):120-124
[5]Li Y W,Yao J H,Liu C J,et al.Int.J.Hydrogen Energy,2010,35(6):2539-2545
[6]ZHUANG Yu-Gui(庄宝贵),LIN Dong-Feng(林东风),CHEN Xiu-Yu(陈秀宇),et al.Chinese J.Inorg.Chem.(Wuji Huaxue Xuebao),2010,26(6):1071-1077
[7]Nethravathi C,Ravishankar N,Shivakumara C,et al.J.Power Sources,2007,172(2):970-974
[8]LIAN Hui-Qin(练慧勤),WANG Jian-Ming(王建明),CHANG Xiao-Tu(常晓途),et al.J.Zhejiang Univ.:Eng.Sci.(Zhejiang Daxue Xuebao:Gongxue Ban),2008,42(7):1223-1226
[9]Gao X R,Lei L X,Hu M,et al.J.Power Sources,2009,191(2):662-668
[10]Demourgues-Guerlou L,Delmas C.J.Power Sources,1993,45(3):2819
[11]Ye X C,Zhu Y J,Zhang Z J,et al.J.Rare Earths,2011,29(8):787-792
[12]Zhang Z J,Zhu Y J,Bao J,et al.J.Alloys Compd.,2011,509(025):7034-7037
[13]ZHANG Zhong-Ju(张仲举),ZHU Yan-Juan(朱燕娟),ZHOU Zhuo-Jun(周焯均),et al.Chinese Patent,CN101982402A.2011-03-02.
[14]Vishnu Kamath P,Dixit M,Indira L,et al.J.Electrochem.Soc.,1994,141(11):29-56
[15]Watanabe K,Kikuoka T,Kumagai N.J.Appl Electrochem.,1995,25(3):219-226
[16]Zhou H B,Zhou Z T.Solid State Ionics,2005,176:1909-1914
[17]Guan X Y,Deng J C.Mater.Lett.,2007,61:621-625
[18]TANG Jin-Hong(唐 金 红),CHEN Shi(陈 实),WANG Fang(王芳),et al.J.Funct.Mater.(Gongneng Cailiao),2007,38(5):696-699
[19]XIE Jing-Ying(解晶莹),SHI Peng-Fei(史鹏飞),ZHOU Ding(周定),et al.J.Harbin Inst.Technol.(Harbin Gongye Daxue Xuebao),1997,29(6):31-33 turn 41
[20]Singh D.J.Electrochem.Soc.,1998,145(1):116-120
[21]Delahaye-Vidal A,Beaudoin B,Sacepee N.J.Solid State Ion,1996,84(3/4):239-24
猜你喜欢
中国食用菌(2021年10期)2021-11-04
新能源汽车供能技术(2021年1期)2021-10-14
发明与创新·小学生(2020年4期)2020-08-14
电子制作(2019年23期)2019-02-23
北京航空航天大学学报(2017年2期)2017-11-24
学生天地(2017年19期)2017-11-06
发明与创新·小学生(2016年4期)2016-08-04
汽车实用技术(2015年8期)2015-12-26
燕山大学学报(2015年4期)2015-12-25
天津科技大学学报(2015年4期)2015-04-16
