
微波辐射下嘌呤C-2,6位衍生物及“Reversine”的合成*
2013-10-16 03:44刘红霞尹志伟王诗玺万升标
中国海洋大学学报(自然科学版) 2013年9期
刘红霞,尹志伟,王诗玺,万升标,江 涛**
(1.中国海洋大学医药学院,海洋药物教育部重点实验室,山东 青岛266003;2.齐齐哈尔大学化学与化学工程学院,黑龙江 齐齐哈尔161006)
嘌呤衍生物是一类重要的抗病毒药物中间体[1],嘌呤环的结构中可以发生取代反应的有2,6,8,9 4个位置,除了嘌呤环的9位接入糖基合成核苷类药物以外,嘌呤母环中可以作为分子修饰的位置还有2,6,8 3个位置。经研究表明,在嘌呤环的2,6或8位碳上引入某些取代基团后,所得的嘌呤衍生物具有抗病毒、抗癌和降血压等重要的生物医学活性[2-4]。因此,嘌呤环的结构修饰一直是人们研究嘌呤类化合物的重点[5,6]。“Reversine”[1,2-(4-吗啉苯胺基)-N6-环己基腺嘌呤]是一种嘌呤的2,6位取代物,研究发现,它是一种高效的人类A3腺苷受体(AR)抑制剂,其Ki值为0.66 μmol/L[7],而且它在辨别白血病等肿瘤细胞方面表现出很好的活性。这些研究结果引起了研究者们的高度重视,进而合成了一系列“Reversine”的衍生物[8],并研究其构效关系。
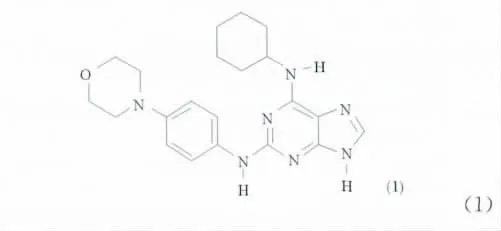
Alise等[9]报道的“Reversine”的合成是以2-氟-6-氯嘌呤为原料,首先在碱性条件下,与环己胺反应,制备2-氟-6-环己胺基嘌呤,再与对吗啉苯胺在乙醇中加压反应制得产物。该方法中第一步6位取代反应要正丁醇中回流24h,第二步2位取代要在乙醇中110℃反应48h,反应时间长,条件要求高。由于嘌呤2位的亲核取代反应比较难以进行,一般制备方法反应时间都比较长或者需要使用特殊的催化剂[10]。Marin T[11]等合成了一系列嘌呤的2,6,9位衍生物,但是其2位的胺取代要150℃,反应30h。Lan R等[12]采用三氟乙酸作溶剂,但2位取代衍生物的反应时间仍然很长。研究者们试图通过改变溶剂、提高温度、提高胺的配比等方法来提高反应的活性[13],但是仍需要数10h的反应时间。为了探索高效的合成嘌呤衍生物及“Reversine”的方法,本文研究以2,6-二氯嘌呤为原料,将绿色的微波辐射方法应用于该反应。实验研究表明,微波能很好的促进嘌呤2,6位取代反应的进行,反应时间被极大的缩短,从而快速的合成了多种新的嘌呤的2,6位胺基取代衍生物及“Reversine”,并研究了反应条件和影响因素,产物的结构通过1H NMR进行了表征。
1 实验部分
1.1 实验仪器与药品
主要仪器 MAS-Ⅱ型常压微波辅助合成/萃取反应仪(上海新仪微波化学科技有限公司);Avance400核磁共振波谱仪(瑞士)。
主要试剂 2,6-二氯嘌呤(南京兆祥工贸有限公司,含量≥99.5%);环己胺、正丁胺、吗啉、哌嗪、苯胺等均为分析纯试剂。
1.2 微波辐射下嘌呤的6位取代衍生物的合成
1.2.1 微波辐射下合成2-氯-6-环己胺基嘌呤 在25mL微波反应器专用反应瓶中加入0.189g(1mmol)的 2,6-二 氯 嘌 呤 (2),N,N 二 异 丙 基 乙胺0.156g(1.2eq)和0.095g(0.9eq)环己胺,安装回流冷凝管,磁力搅拌(545r/min)使混合均匀。设定微波功率100W,设定温度120℃,微波连续照射10min后停止反应。反应液冷却,析出白色固体,抽滤,并用丙酮洗涤滤饼,60℃干燥,得乳白色粉末状固体产物(3a)[14]。1H NMR(DMSO-d6,400MHz)δ:1.434~1.633(m,6H),1.731~1.893(m,5H),4.529(bs,1H,NH),7.978(bs,1H,NH),8.124(s,1H,CH),12.989(bs,1H,NH)。
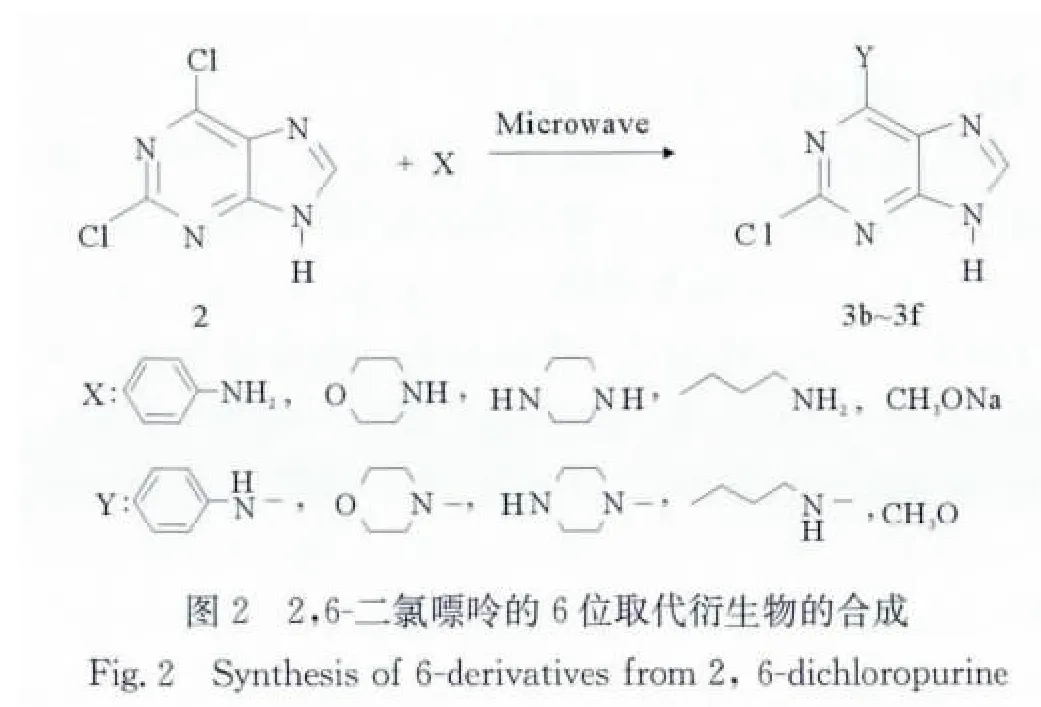
1.2.2 嘌呤6位取代衍生物的合成 用类似的方法,以2,6-二氯嘌呤(2)为原料,在微波辐射下分别与苯胺、吗啉、哌嗪、正丁胺、甲醇钠反应,制备2-氯嘌呤的6位取代衍生物3b~3f。制备2-氯-6-苯胺基嘌呤(3b)时,以1.2eq的三乙胺为催化剂,苯胺配比为1eq,用正丁醇做溶剂,设定功率100W,温度120℃,照射20min。制备3c~3f的方法与1.2.1相同,微波功率100W,设定温度70~120℃,微波连续照射1~15min。
2-氯-6-苯胺基嘌呤(3b):1H NMR(DMSO-d6,400 MHz)δ:7.087(d,J=7.6Hz,1H),7.357(d,J=7.2 Hz,2H),7.841(m,2H),8.285(s,1H,CH),10.194(bs,1H,NH),13.316(bs,1H,NH)。
2-氯-6-(N-吗啉基)嘌呤(3c):1H NMR(DMSO-d6,400MHz)δ:3.708~3.742(m,4H),4.183~4.189(m,4H),8.155(s,1H,CH),13.22(s,1H,NH)。
2-氯-6-(N-哌嗪基)嘌呤(3d):1H NMR(DMSO-d6,400MHz)δ:3.182~3.208(m,8H),4.39(bs,1H),8.22(s,1H,CH)。
2-氯-6-正丁胺基嘌呤(3e):1H NMR(DMSO-d6,400MHz)δ:0.899(t,3H,J=2.8Hz,CH3),1.317~1.334(m,2H,CH2),1.525~1.597(m,2H,CH2),3.399~3.414(m,2H,CH2),3.85(bs,1H,NH),8.09(s,1H,CH),8.17(bs,1H,NH)。
2-氯-6-甲氧基嘌呤(3f):1H NMR(DMSO-d6,400 MHz)δ:4.041(s,3H,OCH3),4.429(bs,1H),8.192(s,1H,CH)。
1.3 2-苯胺基-6-环己胺基嘌呤的合成
在25mL微波反应器专用反应瓶中加入0.255g(1mmol)的2-氯-6-环己胺基嘌呤(3a)和0.93g苯胺(10mmol),浓盐酸催化,安装回流冷凝管,磁力搅拌(533r/min)使混合均匀。微波功率100~300W,TLC跟踪,回流反应20min。反应结束后冷却抽滤,用乙醇洗涤滤饼,60℃烘干,得灰白色粉末状固体(4)。
1H NMR(DMSO-d6,400MHz)δ:1.136~1.412(m,6H),1.631~1.946(m,5H),4.096(bs,1H,NH),6.832(s,1H,ArH),7.205(d,2H,J=7.6Hz,ArH),7.812(d,2H,J=8.4Hz,ArH),8.785(s,1H,CH),12.368(bs,1H,NH)。
1.4 微波辐射下“Reversine”(1)的合成
1.4.1 4-吗啉基苯胺的合成 4-吗啉基苯胺是合成“Reversine”的重要中间体,以4-氟硝基苯为原料制备。100mL三颈瓶中加入2.82g(20mmol)4-氟硝基苯(5)、1.74g(20mmol)吗啉及2.75g(40mmol)碳酸钾,加入溶剂20mL DMSO,90℃搅拌反应8h,反应液冷却后倒入冰水中,过滤,干燥,得黄色固体4-吗啉基硝基苯(6)3.92g(产率94.2%)。4-吗啉基硝基苯1.04g(5mmol),加入乙酸乙酯-甲醇(2∶1)混合溶剂30mL,10%Pd/C 0.01g,N2置换后,通入 H2,室温搅拌反应8h。过滤除去Pd/C,乙酸乙酯洗涤,蒸干滤液,得棕色固体产物 4-吗啉基苯胺 (7)0.767g(产率84.1%)。1H NMR (DMSO-d6,400MHz):δ3.15~3.20(m,4H),3.73~3.89(m,4H),6.60(d,2H),7.11(d,2H)。与文献[15]报道一致。
1.4.2 微波辐射下“Reversine”(1)的合成 25mL微波专用反应瓶中投入2-氯-6-环己胺嘌呤(3a)0.126 g(0.5mmol),4-吗啉基苯胺(7)0.098g(0.55mmol),正丁醇3mL,浓盐酸催化。安装回流冷凝及氮气保护装置,N2置换反应体系。设定温度120℃,微波功率300W,微波照射过程中每10min TLC检测一次,至90min,反应完全。冷却后加入8mL乙酸乙酯,析出固体,抽滤,得到浅棕色的粉末状固体产物“Reversine”(1)。
1H NMR(DMSO-d6,400MHz):(1.145~1.308(m,6H),1.570~1.940(m,5H),2.934~3.077(m,4H),3.531~4.175(m,4H),6.978(d,2H,J=7.2 Hz,ArH),7.544(d,2H,J=3.2Hz,ArH),8.423(s,1H,CH),9.257(bs,1H,NH)。
2 结果与讨论
2.1 微波辐射合成2-氯-6-环己胺基嘌呤的反应条件及影响因素
如图1所示,反应先在100W功率下,70℃下间歇进行,溶剂采用正丁醇,每次微波照射1min,中间进行冷却,TLC跟踪,至30min停止反应,体系产生白色固体。产物收率64%。
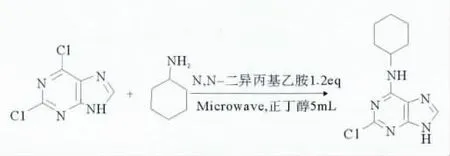
图1 2,6-二氯嘌呤的6位环己胺取代Fig.1 Substituted of 2,6-dichloropurine by cyclohexylamine
在此基础上,为了进一步优化反应条件,又设定温度120℃,采用微波连续照射,反应在回流下进行,分别照射10、15、20min,停止照射后冷却,析出白色固体,产率分别为65.3%,68.2%,69.8%。可见,温度对该反应并无太大的影响,为了方便操作及缩短反应时间,回流状态下连续照射更方便,照射时间10min即可,再延长照射时间对产率影响不大。
另外,对该反应进行了放大,原料2,6-二氯嘌呤增大到10mmol,其他反应条件不变,100W微波连续照射10min,TLC检测反应完全,产物收率可达到86.1%。
2.2 嘌呤6位取代衍生物的合成反应条件及影响因素
在不同的反应条件下,合成2-氯嘌呤的6位取代衍生物3b~3f,反应式如图2所示,反应结果见表1。

?
实验结果表明,2,6-二氯嘌呤与苯胺的反应比较难进行,本文尝试以正丁醇为溶剂,以1.2eq的N,N-二异丙基乙胺为催化剂,苯胺配比0.9~10eq,搅拌反应温度80~120℃,搅拌至10h,或用三乙胺作催化剂(配比为1.2eq),用正丁醇或DMSO做溶剂,80℃搅拌10h,发现均未反应。只有以DMSO为溶剂,120℃反应才有产物生成。
而采用微波辐射方法,以1.2eq的三乙胺为催化剂,苯胺配比为1eq,用正丁醇做溶剂,设定功率100 W,温度120℃,照射20min,由TLC检测发现反应完全,产率71.3%。
2,6-二氯嘌呤与吗啉、哌嗪、正丁胺、甲醇钠的反应,在微波辐射下均能快速的进行,不同的取代反应物所需要的时间有所不同,但均有较高的产率,产物均为白色固体粉末。
2.3 微波法合成6-环己基嘌呤2位取代产物结果
以2-氯-6-环己胺嘌呤为原料,和一定量的苯胺在微波辐射下反应,反应式如图R
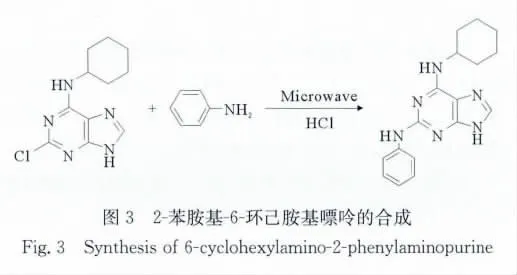
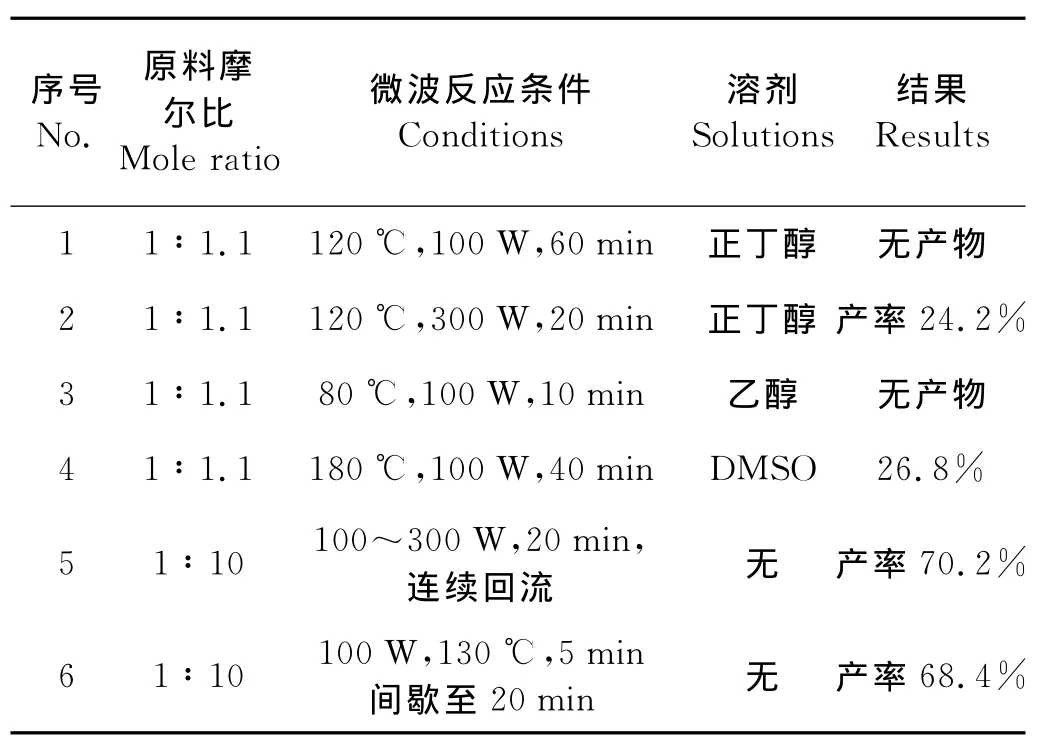
表2 微波法合成2-苯胺基-6-环己胺基嘌呤(4)结果Table 2 Results of the reaction of 2-phenylamino-6-cyclohexylaminopurine(4)by microwave irradiation
由表2可见,微波法制备2-苯胺基-6-环己胺基嘌呤(4),不论是以正丁醇、乙醇或DMSO作溶剂,温度80~180℃,如果原料的配比为1∶1.1,反应时间延长至60min,或是功率提高到300W,反应均不能进行完全,或产率较低。而采用无溶剂,用过量的苯胺反应,不论是130℃间歇微波照射还是在苯胺沸点下回流反应,反应均可以进行完全,收率较高。因此,微波法制备2-苯胺基-6-环己胺基嘌呤的较佳条件是在苯胺过量情况下,微波功率100W,连续微波照射反应20min即可。
2.4 微波辐射下“Reversine”(1)的合成结果
“Reversine”(1)中间体4-吗啉基苯胺反应式如图4所示。微波辐射下“Reversine”(1)的合成如图5所示。
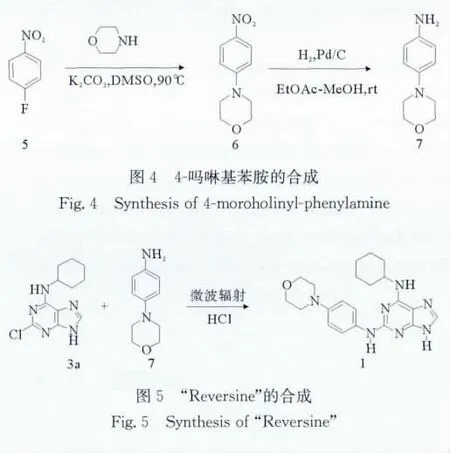
“Reversine”(1)的微波合成比2-苯胺基-6-环己胺基嘌呤(4)容易,在正丁醇溶剂中回流即可反应,而且4-吗啉苯胺的配比采用1∶1.1,由每10min的TLC检测来看,20min时即有产物生成,随着微波照射时间延长至90min时反应完全。产物“Reversine”(1)在正丁醇中有一定的溶解度,但不溶于乙酸乙酯,加入乙酸乙酯后产物析出,过量的4-吗啉基苯胺抽滤时即可除去。由此可见,用微波辅助方法合成“Reversine”(1),反应迅速,产物收率可达70%以上,产物纯度好。
[1] Cai Hancheng,Yin Duanzhi,Zhang Lan,et al.Synthesis of 2-A-mino-6-fluoro-9-(4-hydroxy-3-hydroxymethylbuty1)-purine [J].Chinese Journal of Organic Chemistry,2006,26(12):1709-1713.
[2] Xu Hongyan,Maga Giovanni,Focher Federico,et al.Synthesis,properties,and pharmacokinetic studies of n2-phenylguanine derivatives as inhibitors of herpes simplex virus thymidine kinases[J].J Med Chem,1995,38:49-57.
[3] Estep K G,Josef K A.Synthesis and structure-activity relationships of 6-heterocyclic-substituted purine as inacvation modifiers of cardiac sodium channels[J].J Med Chem,1995,38:2582-2595.
[4] Shum P W,Peet N P,Weintraub P M,et al.The design and synthesis of purine inhibitors of CDK2.III[J].Nucleosides,Nucleotides & Nucleic Acids,2001,20(4-7):1067-1078.
[5] Szczepankiewicz Bruce G,Rohde Jeffrey J,Kurukulasuriya Ravi.Pyrazolo[1,5-a]-1,3,5-triazine as a purine bioisostere:access to potent cyclin-dependent kinase inhibitor (R)-roscovitine analogue[J].J Med Chem,2009,52:655-663.
[6] Manikowski A,Verri A,Lossani A,et al.Inhibition of herpes simplex virus thymidine kinases by 2-phenylamino-6-oxopurines and related compounds:structure-activity relationships and antiherpetic activity in vivo[J].J Med Chem,2005,48:3919-3929.
[7] Anderson Kenneth C,Mitsiades Nicholas,Negri Joseph,et al.Use of reversine and analogs for treatment of cancer:US.,060535[P].2008-05-22.
[8] Perreira M,Jiang Jian-kang,Klutz A M,et al.“Reversine”and its 2-substituted adenine derivatives as potent and selective a3receptor antagonists[J].J Med Chem,2005,48:4910-4918.
[9] Alise A M D,Amabile G,Iovino M,et al.Reversine,a novel Aurora kinases inhibitor,inhibits colony formation of human acute myeloid leukemia cells[J].Molecular Cancer Therapeutics,2008,7(5):1140-1149.
[10] Ding Sheng,Gray N S,Wu Xu,et al.A Combinatorial scaffold approach toward kinase-directed heterocycle libraries[J].J Am Chem Soc,2002,124(8):1594-1596.
[11] Marin T.Fiorini chris abell.solution-phase synthesis of 2,6,9-trisubustituted purines[J].Tetrahedron Letters,1998,39:1827-1830.
[12] Hardcastle Ian R,Arris Christine E,Bentley Johanne,et al.N-substituted O-cyclohexylmethyl-guanine derivatives:potent inhibitors of cyclin-dependent kinases 1and 2[J].J Med Chem,2004,47(15):3710-3722.
[13] Ciszewski Lech,Waykole Liladhar,Prashad Mahavir,et al.A practical synthesis of 2-arylamino-6-alkylaminopurines from 2,6-dichloropurine[J].Organic Process Research & Development,2006,10:799-802.
[14] Huang Ling-Kuen,Cherng Yen-Chih,Cheng Yann-Ru,et al.An efficient synthesis of substituted cytosines and purinesunder focused microwave irradiation[J].Tetrahedron,2007,63:5323-5327.
[15] Tangallapally Rajendra P,Yendapally Raghunandan,Lee Robin E,et al.Synthesis and evaluation of cyclic secondary amine substituted phenyl and benzyl nitrofuranyl amides as novel antituberculosis agents[J].J Med Chem,2005,48(26):8261-8269.
猜你喜欢
能源化工(2022年1期)2023-01-14
含能材料(2022年10期)2022-10-22
广州化工(2022年3期)2022-02-24
能源化工(2021年6期)2021-12-30
纺织检测与标准(2021年3期)2021-12-03
现代信息科技(2021年15期)2021-03-13
世界农药(2020年12期)2021-01-04
农药科学与管理(2019年8期)2019-11-23
火炸药学报(2019年5期)2019-11-11
农药科学与管理(2019年12期)2019-05-20
