
钼/活性炭渣油加氢催化剂的硫化
2013-10-11 02:50:42刘元东
化工进展 2013年8期
刘元东
(中国石油天然气股份有限公司石油化工研究院,北京 100195)
渣油加氢催化剂金属活性组分Mo、Co、Ni、W等通常以氧化态形式存在,在使用前需要经过硫化处理,将金属氧化物转化为硫化物,催化剂才具有优良的加氢活性和稳定性。硫化过程对渣油加氢催化剂的反应性能有重要影响,而影响催化剂硫化效果的因素有硫化温度、硫化时间、硫化氢分压、硫化剂的浓度和种类等,其中,硫化温度对硫化过程影响较大[1-2]。除了硫化操作条件等影响因素外,加氢催化剂活性组分与不同载体间相互作用的强弱对于活性相的分散、形貌、稳定性也有一定的影响。以氧化铝为载体的渣油加氢催化剂由于载体与金属间的强相互作用,使得催化剂的硫化完全有一定困难。近年来,一种以活性炭为载体的新型渣油加氢催化剂以其独特的优势受到研究者的关注[3-4]。研究表明,由于活性炭与活性金属间的弱作用力使得硫化过程易于实现,可以形成更多的有效活性位,有利于提高催化剂的加氢活性[5-7]。因此,深入研究硫化过程中的影响因素,揭示催化剂活性组分与活性炭载体相互作用对于活性相的影响,可以为进一步提高催化剂性能提供有益的借鉴。
一般情况下,催化剂的硫化程度越高,其反应活性和稳定性越好。目前,加氢催化剂的硫化效果一般采用催化剂硫化后的实际吸硫量与催化剂理论吸硫量的比值来表示。通常,加氢催化剂的活性金属不可能完全被硫化,硫化产物中可能存在部分未被硫化的产物,如MoOS化合物等,这些化合物也会结合一部分硫,但是这部分硫化物不是催化剂的活性相,对于提高催化剂的活性无辅助作用。随着分析表征技术的发展,硫化度可以采用XPS技术来表征,以催化剂表面活性金属中代表活性相的Mo4+占总金属Mo的百分数来计算硫化度,可以更加精确地揭示硫化程度和催化剂活性之间的关系。
本文主要考察了硫化剂、硫化介质、硫化时间、硫化温度、硫化压力等因素对钼/活性炭催化剂硫化效果的影响,采用XPS技术分析了催化剂的硫化度,优化了钼/活性炭催化剂硫化的工艺条件,揭示了活性炭和活性金属之间相互作用对活性相的影响,评价了优化硫化条件下催化剂的活性。
1 实验部分
1.1 催化剂制备
钼/活性炭催化剂(Mo/AC)以(NH4)6Mo7O24·4H2O为前体,采用等体积浸渍法制备。
1.2 催化剂硫化
硫化剂的加入量为催化剂前体中所含活性金属元素全部硫化所需硫量的1.2倍。
硫化过程在高压反应釜中进行,模拟程序升温形式对催化剂进行硫化。硫化结束后,将催化剂置于盛有环己烷的广口瓶中保存,确保催化剂与空气隔绝,防止催化剂被氧化。
1.3 催化剂表征
XRD测试在Philips X’Pert Pro系列X射线粉末衍射仪上进行,测试条件:电压40 kV,电流30 mA,扫描范围2θ=10°~90°,扫描速度为4°/min。
SEM采用FEI公司Quanta 200F型扫描电镜,电镜的点分辨率为2 nm,加速电压为20 kV,束斑大小为3.0~4.0。
XPS所用的仪器为美国Thermo Fischer公司生产的VG ESCALAB 250型光电子能谱仪,采用AlKα单色器,分析室真空度小于3.0×10−8Pa。
1.4 催化剂硫化度测定
采用XPS能谱仪扫描硫化样品获得Mo3d能谱,应用分峰软件XPSPEAK 41进行谱图解析,分峰规则如下:①Mo3d轨道自旋分离固定为3.15 eV;②Mo(3d5/2)和 Mo(3d3/2)强度比固定为理论值 1.5;③假设Mo(3d5/2)和Mo(3d3/2)半峰宽(FWHM)相同;④谱图形状分布假设由70%高斯和30%洛伦兹组成。
图1是硫化样品的Mo3d峰能谱图,在229.2 eV和232.4 eV显示出强烈的双重带信号,分别是Mo4+(3d5/2)和Mo4+(3d3/2)的特征结合能位置,说明催化剂有MoS2相生成。在235.8 eV位置出现微弱的Mo6+(3d3/2)的特征峰,在 232.6 eV 处的 Mo6+(3d5/2)谱峰与Mo4+(3d3/2)谱峰重合,说明催化剂中有部分Mo未被硫化,呈现Mo6+的价态。此外,在结合能为226.2eV处还出现了S2s的特征峰,在拟合过程中此峰不能被忽略,否则会使测定结果出现偏差。根据以上各峰对应的不同结合能位置,对Mo3d峰进行分峰拟合处理[8],计算出硫化度SXPS如式(1)。

1.5 实验原料性质
实验中采用的某常压渣油的性质如表1所示。


表1 实验原料的性质
1.6 催化剂性能评价
在高压釜中模拟渣油加氢反应,催化剂加入量以活性金属氧化物的质量与原料的质量之比表示,单位为μg/g。评价参数详细含义如式(2)~式(6)所示。
2 结果与讨论
2.1 硫化剂的选择
硫化剂的作用是在硫化过程中提供硫化氢与金属氧化物反应,最基本的硫化剂是硫化氢。实验中采用硫粉(S)和二硫化碳(CS2)作为硫化剂,考察了两种硫化剂的硫化效果。
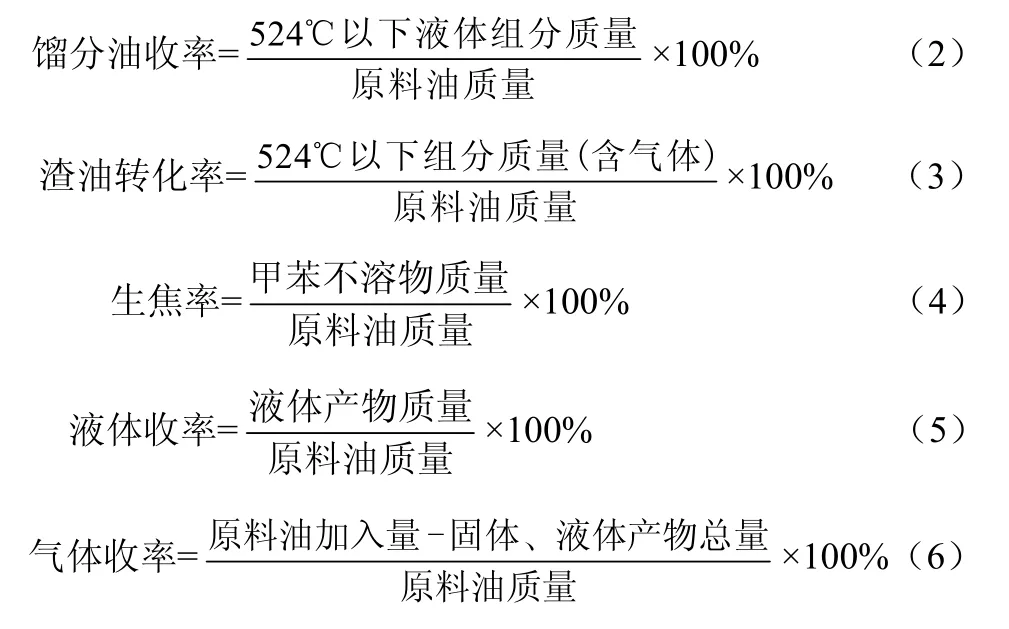
从表2可以看出,CS2和S的硫化效果是不同的,其中,采用CS2硫化后的样品硫化度是71.9%,比S单质的硫化效果好。硫化过程中,采用CS2硫化时,硫化前样品在CS2溶液中浸渍一段时间有利于部分CS2在活性炭孔隙结构中的吸附。硫化过程中,被吸附的CS2与H2反应生成H2S,促进了活性炭内表面活性组分的硫化;采用硫粉硫化时,硫粉与H2生成的H2S只能通过扩散进入催化剂孔隙内,导致内表面的活性组分的硫化程度比CS2低。
图2是硫化前和经过不同硫化剂硫化后的XRD谱图,从中可以看出,未经硫化的氧化态样品Mo/AC在2θ为12.3°、25.7°和30.3°处出现了MoO3特征峰。CS2硫化后样品(Mo/AC/CS2)和S硫化后样品(Mo/AC/S)谱图基本一致,两者均没有出现MoO3或者MoS2的特征衍射峰,只有在2θ为25.0°和44.0°处出现活性炭弱化的特征衍射峰,说明两种硫化方法制备的硫化物种在活性炭上分布比较均匀。

表2 不同硫化剂的硫化效果比较
2.2 硫化介质的选择
加氢催化剂的硫化是一个放热过程,为了避免硫化过程出现局部高温导致硫化不均匀,需要加入合适的硫化介质取热。硫化介质的选择既要考虑有利于催化剂硫化过程的取热,又要方便催化剂硫化完后的分离。实验中以CS2作为硫化剂,比较了润滑油基础油和正十六烷两种不同分散介质对硫化效果的影响,结果如表3所示。
从表3可以看出,采用润滑油基础油作为分散介质硫化度相对较低,采用正十六烷作为硫化介质硫化度在80%以上。硫化过程中,H2S需要通过分散介质扩散至催化剂表面与活性组分发生硫化反应,与正十六烷相比,润滑油黏度和密度较大,扩散传质效果低于正十六烷;同时,在硫化后的分离操作中,需要反复使用甲苯抽提洗去分散介质,由于润滑油稠度较大,抽提和干燥时间较长,有可能暴露在空气中导致活性相被空气氧化从而使硫化效果变差。正十六烷密度和黏度较小,反应完毕后可以使用离心分离快速地将其与催化剂分离,避免了在空气中被氧化。

表3 不同硫化介质的硫化效果比较
图3是在不同硫化介质中硫化的催化剂在相同放大倍数下的SEM图。从图3中可以看出,采用正十六烷作为硫化介质的催化剂S-hexadecane颗粒分散比较均匀,相互之间没有粘连现象;采用润滑油作为硫化介质的催化剂S-sulfur颗粒相对较大,有些颗粒间相互连接,可能是催化剂硫化完后在分离过程中部分被空气氧化发生了轻微的团聚。
2.3 硫化温度对硫化效果的影响
硫化温度是影响加氢催化剂硫化反应过程的重要操作参数,在硫化反应过程中,如果温度过低,则会影响反应速率,延长硫化时间;如果温度过高,金属氧化物在氢气存在条件下以还原反应为主,有可能还原为低价氧化物或金属。低价氧化物与金属再与硫化氢反应时,生成硫化物的反应速率很慢。实验考察了不同硫化温度对硫化度的影响。

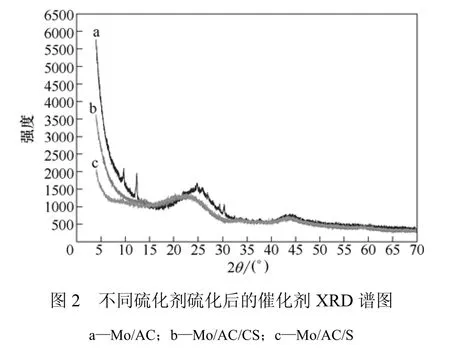
图4是硫化温度对硫化度的影响趋势曲线。在硫化温度280~370℃范围内,硫化度随着温度的升高而呈现出逐渐增加的趋势,当硫化温度到达370℃后,硫化度SXPS达到82%。在一定程度上,利用XPS强度比IS2p/IMo3d变化可以反映出硫化度的变化趋势。图5是XPS强度比IS2p/IMo3d与硫化温度的关系曲线。在280~370℃范围内,XPS强度比IS2p/IMo3d随着硫化温度增加而增大,说明在硫化温度对催化剂硫化效果影响方面,采用XPS强度比IS2p/IMo3d和SXPS结合的方式得到的趋势一致。硫化反应是放热反应,在较低的温度下有利于反应平衡向右移动,但是如果温度过低,就会导致硫化速率太慢,不利于催化剂硫化度的提高。适当提高硫化温度,可以加快硫化反应速度,提高催化剂硫化度,但是温度过高会造成催化剂的局部烧结而降低催化剂的活性。实验中所用高压反应釜温度控制精度为±3℃,为了保证硫化反应速度,同时避免催化剂烧结,选择350℃作为硫化终温。
2.4 硫化时间对硫化效果的影响
加氢催化剂硫化时间不能过短,否则会导致催化剂硫化不完全,降低催化剂活性;如果硫化时间过长,虽然可以小幅度增加硫化度,但是超过平衡极限值后,再延长硫化时间没有实际意义。
图6是硫化度随硫化时间变化的趋势图。从图6中可以看出,随着硫化时间的增加,催化剂的硫化度呈现逐渐提高的趋势,当硫化时间超过2 h后,硫化度的变化幅度相对较小,趋于平缓。在硫化反应开始的2 h内,催化剂硫化度的增加是比较快的,硫化度从62%增加到80%。事实上,每个温度下都有一个平衡极限值,即使再延长硫化时间,硫化度的提高余地也不大,在当前的实验条件下,硫化时间3 h可以确保催化剂硫化度为81%。
2.5 氢气初压对硫化效果的影响

图7是不同硫化压力对催化剂硫化度的影响曲线图。在氢气初压小于5 MPa时,提高压力对于硫化度的增加不明显,当氢气初压从5 MPa提高至6 MPa时,硫化度的提高速度很快,当氢气初压超过6 MPa后,再增加压力,硫化度的变化处于一个相对平缓的阶段。对于液相硫化来说,硫化压力是通过影响催化剂表面积炭状况及硫化介质的相态而影响硫化效果的。在液相硫化过程中,硫化介质在压力较低而温度较高时可能会发生缩合反应甚至生焦,这可能是导致催化剂在5 MPa以下硫化效果不理想的主要原因。随着氢气初压的提高,硫化过程中H2的浓度也会增加,可以有效抑制积炭的生成,从而有利于催化剂硫化度的提高。因此,在较低压力下应选择较低的硫化温度,只有达到一定硫化压力,足够抑制硫化介质的积炭反应时,才可以选择较高的硫化温度。
2.6 硫化态催化剂的表征
通过对钼/活性炭催化剂的硫化规律的研究,总结出较佳的硫化条件:硫化剂采用CS2,硫化介质采用正十六烷,硫化终温350℃,硫化时间3 h,氢气初压6 MPa。在优化条件下对钼/活性炭催化剂进行硫化,硫化后催化剂的硫化度SXPS为85%。通过SEM、TEM等表征手段对硫化态催化剂的结构、形貌、活性相等进行分析,揭示硫化态催化剂的结构特性与催化剂活性之间的关联。
2.6.1 硫化态催化剂的形貌
图8是氧化态和硫化态的催化剂放大10 000倍的SEM图片。从图8中可以看出,未经硫化的催化剂CAT-US表面颗粒大小不一,大部分颗粒粒径很小,有一小部分颗粒粒度相对较大;硫化后的催化剂CAT-S表面颗粒分布比较均匀,粒子直径变小,无大颗粒聚集的情况发生。
2.6.2 硫化态催化剂的活性相
图9为硫化态催化剂MoS2/AC的TEM照片。图中细长的条纹状黑线代表着MoS2晶片结构,单一的黑线对应着单层的MoS2结构,多条平行黑线则代表着多层的MoS2结构。从图9中可以看出,MoS2晶粒呈典型的平行层状结构,平行黑线的层间距为0.61nm左右,是典型的体相MoS2晶面间距的大小,代表着MoS2的(003)晶面[9-10]。活性相MoS2晶粒比较均匀地分散在活性炭载体的表面,没有发现较大MoS2晶粒的聚集。
通常采用堆积层数和晶片长度两个参数考察活性相MoS2晶簇的性质,从图9可以看出,所制备的MoS2/AC硫化态催化剂具有很高的MoS2堆积层数,多为4~6层,同时晶片长度在6~10 nm之间。研究表明[11-12],传统负载型Ni(Co)-Mo/A12O3催化剂的MoS2堆积层数主要以单层和双层为主,且MoS2晶片长度主要分布在2~4 nm之间,这种低的MoS2堆积层数往往会导致活性组分形成催化活性较低的Ⅰ类活性相。相比之下,MoS2/AC催化剂则具有较高的MoS2堆积层数,有利于降低活性金属的空间位阻,提高金属的利用率。同时,由于活性炭载体与活性组分间不存在强烈的相互作用,导致活性组分形成催化活性较高的Ⅱ类活性相,有效地提高催化剂的加氢活性[13]。

2.7 硫化态催化剂的活性评价
采用优化条件下的硫化态催化剂,以某常压渣油作为原料,催化剂加入量800 μg/g,在反应温度420℃,氢气初压8.0 MPa,反应时间60 min条件下进行加氢反应,考察硫化态催化剂的加氢反应效果,结果见图10。

从图10中可以看出,渣油加氢反应过程中气体收率和结焦率均处于比较低的水平,分别为4.3%和1.2%;同时,液体收率、渣油转化率、馏分油收率则分别达到了94.5%、77.1%和72.8%,说明硫化态催化剂具有良好的加氢活性。
3 结论
(1)考察了不同硫化剂和硫化介质的硫化效果,优化了催化剂硫化条件:以CS2为硫化剂,正十六烷为硫化介质,硫化终温350℃,硫化时间3 h,氢气初压6 MPa。在此条件下催化剂硫化度SXPS为85%,MoS2堆垛的片层结构分布比较均匀,数量较多,具有良好的加氢活性。
(2)以硫化度为指标,考察了硫化时间、硫化温度、硫化压力等因素对催化剂硫化效果的影响规律。硫化度随硫化温度的升高而逐渐增加,温度超过350℃后,硫化度随温度的增加提升的幅度很小;随着硫化时间的增加,硫化度逐渐提高,当硫化时间超过3 h后,硫化度达到平衡极限值;氢气初压小于6 MPa时,压力提高,硫化度增加明显,超过6 MPa后,硫化度变化平稳。
(3)活性组分与活性炭间无强烈的相互作用,易于形成催化活性较高的Ⅱ类活性相,活性相MoS2堆垛结构为4~6层,晶片长度为6~10 nm,可以有效地提高催化剂的加氢活性。
[1]李大东.加氢处理工艺与工程[M].北京:中国石化出版社,2004:66-67.
[2]徐春明,杨朝合.石油炼制工程[M].北京:石油工业出版社,2009:398-400.
[3]刘元东,宗保宁,赵愉生,等.活性炭负载型催化剂的制备及其在渣油加氢中的应用[J].化工进展,2011,30(10):2209-2214.
[4]刘元东.钼/活性炭渣油加氢催化剂的制备[J].化工进展,2012,31(12):2708-2709.
[5]Sayag C,Benkhaled M,Suppan S,et al.Comparative kinetic study of the hydrodenitrogenation of indole over activated carbon black composites(CBC)supportedmolybdenum carbides[J].Applied Catalysis A:General,2004,275(1-2):15-24.
[6]Matos J,Laine J.Ethylene conversion on activated carbon-supported NiMo catalysts:Effect of the support[J].Applied Catalysis A:General,2003,241(1-2):25-38.
[7]Suppan S,Trawczynski J,Kaczmarczyk J,et al.Effect of carbon black composite(CBC)support properties on hydrodesulfurization performance of sulfided Mo and Co,and carburized Mo,catalysts[J].Applied Catalysis A:General,2005,280(2):209-214.
[8]Qiu L,Xu G.Peak Overlaps and corresponding solutions in the X-Ray photoelectron spectroscopic study of hydrodesulfurization catalysts[J].Applied Surface Science,2010,256(11):3413-3417.
[9]Ferdous D,Dalai A K,Adjaye J,et al.Surface morphology of NiMo/Al2O3catalysts incorporated with boron and phosphorus:Experimental and simulation[J].Applied Catalysis A:General,2005,294(1):80-91.
[10]Huirache-Acuña R,Albiter M A,Espino J,et al.Synthesis of Ni-Mo-W sulphide catalysts byex situdecomposition of trimetallic precursors[J].Applied Catalysis A:General,2006,304:124-130.
[11]Berhault G,Rosa de la M P,Mehta A,et al.The single-layered morphology of supported MoS2-based catalysts—The role of the cobalt promoter and its effects in the hydrodesulfurization of dibenzothiophene[J].Applied Catalysis A:General,2008,345:80-88.
[12]Li M,Li H,Jiang F,et al.Effect of surface characteristics of different alumina on metal–supportinteraction and hydrodesulfurization activity[J].Fuel,2009,88:1281-1285.
[13]Li G,Li W,Zhang M,et al.Morphology and hydrodesulfurization activity of CoMo sulfide supported on amorphous ZrO2nanoparticles combined with Al2O3[J].Applied Catalysis A:General,2004,273(1-2):233-238.
猜你喜欢
石油沥青(2023年5期)2023-12-08 08:35:04
石油炼制与化工(2023年1期)2023-02-07 09:38:06
黄金(2021年4期)2021-09-10 07:22:44
石油沥青(2019年4期)2019-09-02 01:41:56
中国特种设备安全(2019年3期)2019-04-22 05:05:38
山东工业技术(2018年15期)2018-09-26 10:05:20
石油炼制与化工(2018年5期)2018-03-23 09:04:18
科技创新与应用(2017年4期)2017-03-27 15:22:04
山东工业技术(2016年15期)2016-12-01 05:30:43
橡胶科技(2016年7期)2016-07-28 10:10:52
