
金属氧化物纳米复合催化剂的研究进展
2013-10-11 02:50范立群隋吴彬
化工进展 2013年8期
范立群,隋吴彬
(枣庄学院化学化工与材料科学学院,山东 枣庄277160)
过渡金属的氧化物是一类很重要的催化剂,通过改变其粒径尺寸、粒子形状等提高其催化活性的研究多有报道[1-5]。金属氧化物粒子可以通过溶剂热法、溶胶-凝胶法和浸渍法等方法制备。此外,金属氧化物的形状可通过调节作为诱导剂的表面活性剂的比例而改变。图1是采用不同活性剂浓度制备出的子弹状、钻石状、竿状和分支状的TiO2透射电镜(TEM)照片[6]。据报道,很多金属氧化物如TiO2、Al2O3和ZnO表面既具有路易斯酸又有路易斯碱,使之对很多有机化合物具有良好的吸附性能[4]。金属氧化物高的表面积、体积比、良好的稳定性和可循环使用性,使之成为各种有机化学反应催化剂的不二选择[5]。
1 金属氧化物纳米复合催化剂
1.1 二氧化钛

在众多的金属氧化物中,TiO2由于能吸收紫外光一直以来被视为活性很好的光催化剂[7-8],且通过引入合适的掺杂剂可使其吸收波长延伸到可见光,研究表明,掺杂氮元素的TiO2光催化粒子吸光范围可达可见光[9]。Zhang等[10]通过湿化学法制备出了粒径可控的自由分散的TiO2纳米粒子,系统研究了纳米TiO2或掺杂纳米粒子的粒径大小对光催化活性的影响。纳米粒子粒径大小是一个重要的参数,直接影响催化剂的比表面积。具有较小颗粒尺寸的纳米粒子表面活性位数量大,表面载流子的传输速率也大,光催化活性较强。然而,以往的研究表明,光催化效率不单纯随着粒径减小而增加,而是存在一个最佳粒径。在液相光催化降解氯仿中,TiO2纳米粒子的最佳颗粒大小约10 nm,这是由于增大的表面电子-空穴再复合效应抵消了纳米晶TiO2极高比表面积的催化活性。纳米TiO2的电子-空穴重组可以被分成两类:体积复合和表面复合。体积复合主要发生在结晶良好的大颗粒TiO2中,并可通过减小粒径降低其发生概率,同时减小粒径导致其比表面积增加,因而形成了大量表面活性位,最终产生高的表面载流子传输效率,从而提高催化活性。然而,当纳米晶半导体粒子粒径小到几个纳米时,表面复合成为影响催化活性的主要因素。对于超细粒径纳米材料产生的电子-空穴对及其接近表面从而能迅速到达表面,表面存在空穴和缺乏分离电子-空穴对复合的外力而极易发生表面复合。由于表面电子-空穴重组时间比界面载流子传输过程快得多,对于无限减小的纳米粒子,增加界面传输速率远比增加表面重组复合率重要得多。实验结果表明,纳米TiO2粒子的催化活性随着粒径由21 nm减小到11 nm而增加,粒径进一步减小到6 nm时其催化活性反而降低,因此纳米TiO2光催化体系存在一与最高催化活性相对应的最佳粒径。Zhang等[10]通过实验证实,在液相体系中,TiO2光催化降解CHCl3的最佳粒径是10 nm。
减少电子-空穴重组可提高纳米晶光催化剂的量子产率,通过选择金属离子掺杂到TiO2晶格缺陷中能有效降低电子-空穴的重组。通过研究TiO2粒子尺寸效应和掺杂Fe3+光催化降解效应,发现影响掺杂TiO2催化剂活性的因素与掺杂剂Fe3+的浓度和TiO2粒径大小有关,随着TiO2粒径减小,掺杂剂Fe3+浓度增高,催化活性增强。这是因为在理想状态下电子-空穴对到达界面前被Fe3+捕获,当TiO2粒径变大时,电子-空穴对到达界面的时间延长,如不降低Fe3+的浓度将产生大量的电子-空穴对被捕获,从而降低催化活性。Fe3+掺杂纳米TiO2在光催化降解氯仿反应中表现出比Degussa P25更高的光催化活性。掺杂元素通过与金属纳米粒子形成复合物可有效控制TiO2纳米粒子的光诱导电荷分离,不同平均粒径纳米TiO2的TEM和高分辨率TEM(HRTEM)照片如图2所示。
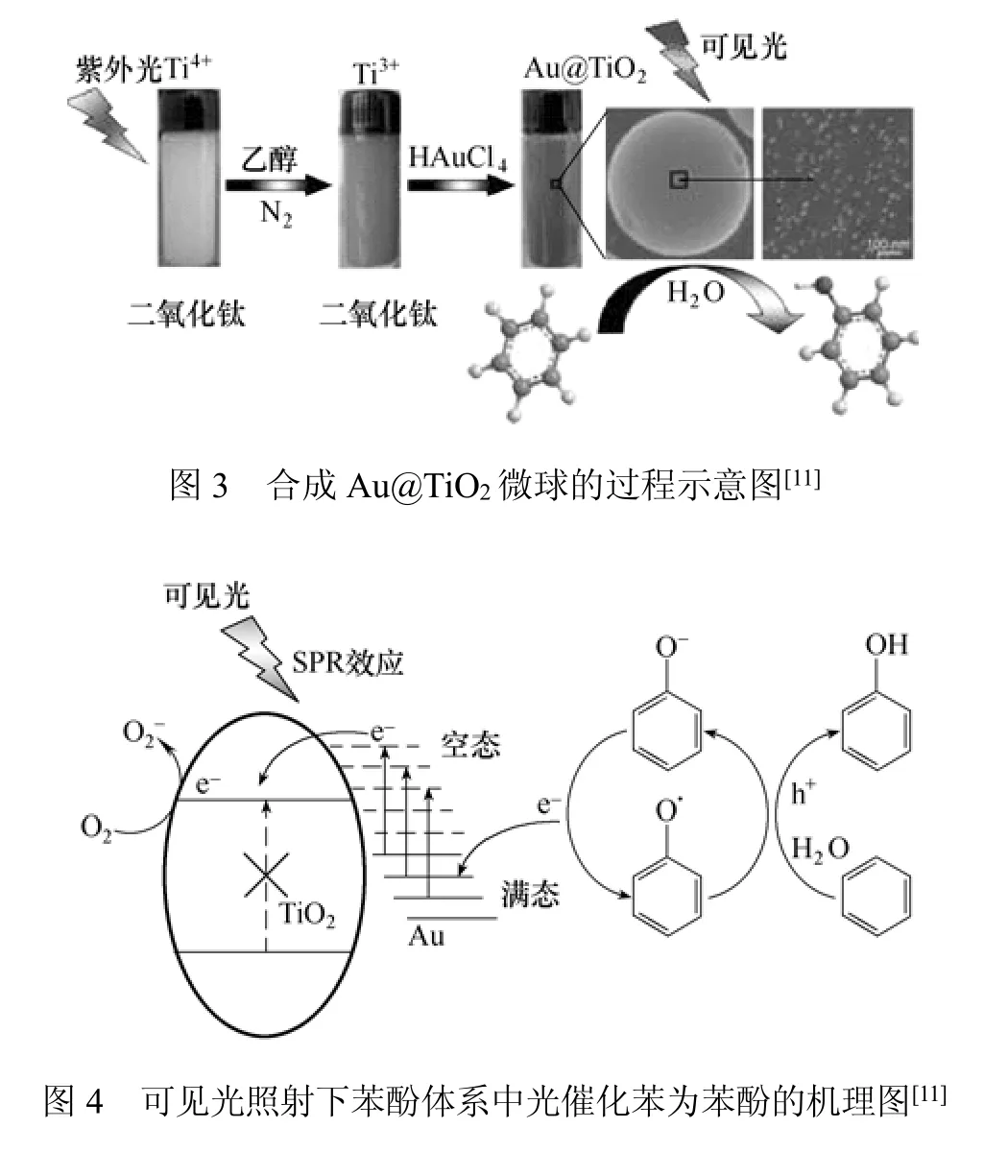
Huang等[11]制备了电浆光催化金-TiO2复合催化剂,合成Au-TiO2复合纳米粒子的过程原理如图3所示。通过在可见光下Au@TiO2光催化氧化苯考察了其催化活性,结果表明,Au@TiO2复合催化剂比Pt@TiO2和Ag@TiO2复合催化剂表现出更高的光催化活性,这主要是由于前者中缺电子的金将苯氧基阴离子氧化为自由基粒子,极大促进了苯氧化为苯酚。推测其可能机理为:在可见光照射下,Au@TiO2中Au纳米粒子的光激发电子快速穿过Au-TiO2界面到达TiO2的最低导带。因此,在酚水溶液中,悬浮Au@TiO2复合物经可见光照射,诱发电子从Au纳米离子到TiO2粒子的迁移被溶液中同时发生的从酚到Au纳米离子的电子转移所抵消。该结论为电子顺磁共振(EPR)和Au的X射线光电子能谱分析(XPS)检测结果所验证,机理如图4所示。
1.2 赤铁矿
赤铁矿(α-Fe2O3)基于氧与铁在八面体空穴2/3处六方紧密堆积结构特征,作为催化剂已被广泛研究,由于其低成本、高抗腐蚀、环境友好等特点,广泛用作气体传感器和电极材料等。良好的应用前景促进了对各种纳米结构α-Fe2O3材料制备方法的研究,现已提出了热分解、热氧化和水热合成法等方法。Zheng等[1]首次利用硝酸铁作为金属离子源在聚乙烯吡咯烷酮(PVP)体系中合成制备出了单分散、单晶、晶粒大小为40 nm的准立方形α-Fe2O3纳米粒子,且形貌可通过改变合成制备参数调控。所制备的准立方形α-Fe2O3纳米催化剂的活性由催化氧化CO来验证,其在催化反应过程中的活化温度、转化效率和热稳定性等特征表明,其催化活性远远优于其它形式的纳米或微小尺寸氧化铁催化剂。通过溶剂热法合成制备了6个相同{110}晶面的准立方形α-Fe2O3纳米粒子,结果表面活性剂聚乙烯吡咯烷酮(PVP)对制备最终产品形貌结构起重要作用。表面活性剂PVP不仅是α-Fe2O3纳米粒子的稳定剂和分散剂,还控制形成的准立方形的几何形态,如果没有PVP,大部分纳米粒子被截断或者不能形成准立方形构型;随着PVP加入量的增加,α-Fe2O3纳米粒子附着在PVP的长碳链上,聚集速率缓慢,使其具有更长的时间选择最优的空间位置而最终形成准立方形结构。

类似贵金属和半导体催化剂,α-Fe2O3的催化活性强烈地依赖于颗粒的形状,且可以通过控制前体的比例和反应时间改变颗粒的形状。以准立方形α-Fe2O3为催化剂,在230℃下可将CO完全氧化成CO2,而相同实验条件下,花型α-Fe2O3只能将不到5%的CO氧化为CO2。α-Fe2O3纳米粒子有6个当量晶面{110}含有较高密度的Fe原子,CO首先被吸附在Fe的表面,随后被氧化成CO2,氧原子的协同氧化作用较小。准立方体α-Fe2O3表现出比花型、中空型或其它形式的不规则晶面更高的催化活性,是由于准立方体型具有更多的具有活性Fe原子的{110}晶面。不同形状的α-Fe2O3纳米粒子的TEM照片如图5所示。
1.3 氧化锌
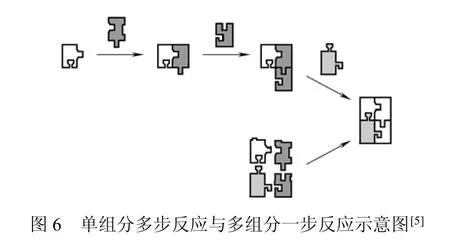
多组分反应是有机合成领域一种新兴的有效反应方法,在一步反应过程中生成多种新的化学键。众所周知,在多步法合成过程中,反应和纯化的次数可用于评价该反应的有效性和实用性,而且操作越简单越好。单组分多步合成法与多组分一步合成法如图6所示。

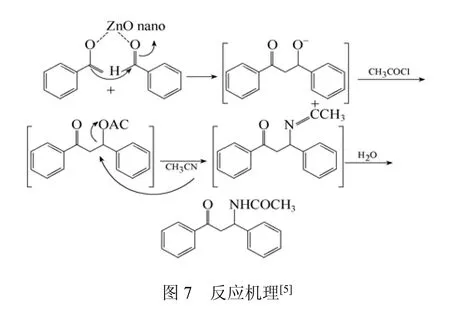
ZnO是价格低廉、环境友好的多组分一步法反应催化剂[5,9],其在合成化学和制药工业中重要化合物β-乙酰氨基酮/酯的反应中表现出良好的催化活性。β-乙酰氨基-β-苯基苯丙酮的合成在室温下进行,在以ZnO为催化剂的反应体系中,ZnO的摩尔百分比为10%~20%时反应产率可增加到88%;而在相同反应体系中不添加任何催化剂的情况下,反应30 h只有5%的产率;而在ZnO摩尔比为10%的体系中,反应1 h产率就可达4%。推断其催化反应机理一方面可能是纳米粒子ZnO协同醛上的氧原子激活苯甲醛亲核攻击的活性(图7);另一方面,ZnO纳米粒子具有较高的比表面积使其具有更强的催化活性,更有利于苯乙酮的烯醇化[5]。
此外,纳米ZnO的高催化活性还表现在其对乙醇和苯酚的O-酰化反应的催化作用。Firouz和Hamdollah[9]利用微波辅助溶剂热法由Zn(CH3CO2)2·2H2O 与NaOH合成制备出了粒径为30 nm的ZnO粒子,SEM和TEM表征结果表明,ZnO纳米粒子具有球形结构。从制备过程中影响ZnO粒径大小的因素来看,反应时间和溶剂的去除对ZnO粒径具有重要影响。除了纳米ZnO颗粒外,块状ZnO催化乙醇和苯酚的O-酰化反应产率可达到65%,对比不添加任何催化剂的相同反应体系,在反应长达240 min后仍未观察到有反应发生,而用纳米ZnO颗粒作催化剂的反应体系的反应时间是用块状ZnO的1/24倍,产率高达92%。反应机理如图8所示。
1.4 氧化钨

金属氧化物粒子如纯的或掺杂的TiO2、WO3可有效分解挥发性有机化合物(VOC)。由于WO3的导带水平(+0.5 V)比O2的氧化还原水平更正,长期以来被视为不适合催化降解空气中有机物和具有很强电子受体的反应体系,而掺杂Pt的WO3在可见光照射下降解有机化合物的能力显著提高。Abe等[13]利用光沉积法用H2PtCl6·6H2O作Pt源,在纯水体系中,在有可见光照射的条件下将Pt负载到WO3微球上,而后转移到甲醇溶液中,Pt得以高度均匀分散负载到WO3微球上。WO3粒子与Pt-WO3粒子的TEM照片如图9、图10所示。在可见光照射下评价Pt-WO3降解有机物的催化活性,并与在相同实验条件下N-TiO2和 WO3同时降解乙酸、乙醛的结果进行对比,结果表明Pt-WO3的催化活性显著高于N-TiO2和 WO3,其催化活性甚至可与在紫外光下的TiO2的催化活性相提并论。
利用光声光谱(PA)测定掺杂Pt的WO3在反应过程中的电子转移机理,惊奇地发现WO3中产生的光激发电子更倾向于与O2发生反应,且Pt的存在增强了这种反应效应[13]。掺杂Pt后WO3催化活性的提高归因于Pt的存在促进了多电子还原而非单电子还原。该研究表明,利用简单氧化物负载纳米离子的复合催化剂促进多电子还原O2,从而显著提高可见光下高催化活性和耐用性催化剂的策略是行之有效的[12-14]。
1.5 氧化亚铜
与贵金属催化剂相似,金属氧化物纳米粒子的催化性能可通过制备特殊晶型结构和裸露晶面来控制。氧化亚铜(Cu2O)的催化活性取决于其晶型结构在CO的氧化反应中得到了验证。o-Cu2O(八面体Cu2O)、c-Cu2O(立方体Cu2O)、CuO/o-Cu2O和CuO/c-Cu2O催化氧化CO(图11),实验结果表明,其它3种晶型结构均比c-Cu2O表现出优越的催化活性。此外,发现o-Cu2O和CuO/o-Cu2O分别在150℃和240℃下具有好的催化活性,在240℃CO转化率的高达92%;而在190℃反应温度下c-Cu2O和CuO/c-Cu2O催化氧化CO,其转化率均低于50%。就CO的氧化,对Cu2O{111}和 {100}晶面所做的密度函数理论计算表明,由于反应中间产物的活化能不同(分别为0.37 eV和 1.15 eV),催化发生在CuO催化活性薄膜上且不同的晶面催化活性不同。这些实验结果证明,独特的催化活性可以在氧化物纳米粒子特定的结晶面来实现,并可通过控制晶面的生长进一步提高。

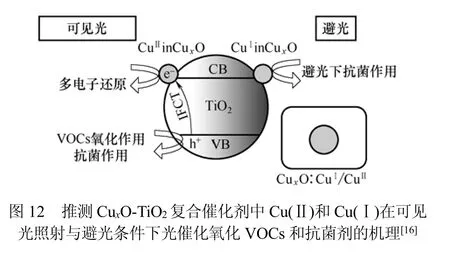
最近,在CuxO与TiO2组成的复合纳米催化材料中,Ⅰ价Cu具有很高的抗菌活性,Ⅱ价Cu在可见光下对VOC具有很高的光催化氧化活性[16]。除了高活性Cu2O-TiO2复合纳米催化剂外,纳米团簇CuxO与TiO2组成的复合纳米催化材料具有降解室内VOC催化活性和抗菌活性这一现象表明,在CuxO-TiO2复合催化剂中,TiO2是活性物质,Cu(Ⅱ)有效提高了TiO2在可见光下的光催化活性,同时在界面间电子转移过程中TiO2价带电子激活Cu(Ⅱ)电子,被激活的电子通过多电子还原有效减少氧分子。除此之外,价带上具有较强氧化能力的空穴结构有效降解有机物质[13]。纳米复合材料的抗菌活性可利用光催化抗菌作用的标准方法观察噬菌斑测定,其中包括能够感染大肠杆菌的噬菌体Qβ测试解决方案。对比可见光和避光条件下0.25%Cu(Ⅱ)/TiO2催化失活噬菌体发现,其在可见光下催化活性显著,避光下具有微弱的催化活性。虽然纳米复合催化剂的活性在避光下超过I价Cu,当光照射类似组成的纳米复合材料时,在短时间内即能获得很高的催化活性。推测CuxO/TiO2中Cu(Ⅱ)和Cu(Ⅰ)在可见光照射与避光条件下光催化氧化VOCs和抗菌剂的机理见图12。
此外,辐照CuxO/TiO2纳米复合材料发现,复合材料中存在过量的Ⅰ价Cu有更好的抗病原体活性。Ⅰ价Cu随后产生由TiO2到Ⅱ价Cu的界面电子转移,同时界面电子转移到TiO2的空穴价带中。这两种电子迁移显著提高了其抗病原体活性。
2 总结与展望
通过总结金属氧化物及复合双金属纳米粒子粒径的大小、形状和暴露在反应体系中的不同晶面对不同催化体系催化效率的比较以及产品产量高低的关系看出,设计、合成制备高效实用的金属纳米复合催化材料是促进有机化学领域与光催化氧化领域飞速发展的有效途径,同时也是近年来纳米催化领域飞跃发展的必然趋势。
[1]Zheng Y,Cheng Y,Wang Y,et al.Quasicubic α-Fe2O3nanoparticles with excellent catalytic performance[J].Phys.Chem.B,2006,110:3093-3097.
[2]Radwan N R E,El-Shall M S,Hassan H M A.Synthesis and characterization of nanoparticle Co3O4,CuO and NiO catalysts prepared by physical and chemical methods to minimize air pollution[J].Applied Catalysis A:General,2007,331:8-18.
[3]Irie H,Miura S,Kamiya K,et al.Efficient visible light-sensitive photocatalysts: Grafting Cu(Ⅱ) ions onto TiO2and WO3photocatalysts[J].Chem.Phys.Lett.,2008,457:202-205.
[4]Tanabe K.Solid Acids and Bases[M].New York:Academic Press,1970.
[5]Mirjafary Z,Saeidian H,SadeghiA,et al.ZnO nanoparticles:An efficient nanocatalyst for the synthesis of β-acetamido ketones/estersviaa multi-component reaction[J].Catalysis Comm.,2008,9:299-306.
[6]Kudera S,Carbone L,Casula M F,et al.Selective growth of PbSe on one or both tips of colloidal semiconductor nanorods[J].Nano Lett.,2005,5:445-449.
[7]Linsebigler AL,Lu G,Yates Jr J T.Photocatalysis on TiO2surfaces:Principles,mechanisms,and selected results[J].Chem.Rev.,1995,95:735-758.
[8]KubackaA , Fernández-GarcíaM , Colón G.Advanced nanoarchitectures for solar photocatalytic applications[J].Chem.Rev.,2012,112:1555-1614.
[9]Firouz M M,Hamdollah S.Controlled microwave-assisted synthesis of ZnO nanopowder and its catalytic activity forO-acylation of alcohol and phenol[J].Sci.and Eng.B,2007,139:265-269.
[10]Zhang Z,Wang C,Zakaria R,et al.Role of particle size in nanocrystalline TiO2-based photocatalysts[J].Phys.Chem.B,1998,102:10871-10878.
[11]Zheng Z,Huang B,Qin X,et al.Facilein situsynthesis of visible-light plasmonic photocatalysts M@TiO2(M=Au,Pt,Ag)and evaluation of their photocatalytic oxidation of benzene to phenol[J].Mater.Chem.,2011,21:9079-9087.
[12]Papaefthimiou P,Ioannides T,Verykios X E.Performance of doped Pt/TiO2(W6+)catalysts for combustion of volatile organic compounds(VOCs)[J].Applied Catalysis B:Environmental,1998,15:75-92.
[13]Abe R,Takami H,Murakami N,et al.Pristine simple oxides as visible light driven photocatalysts:Highly efficient decomposition of organic compounds over platinum-loaded tungsten oxide[J].Am.Chem.Soc.,2008,130:7780-7781.
[14]Liu Y,Xie C,Li J,et al.New insights into the relationship between photocatalytic activity and photocurrent of TiO2/WO3nanocomposite[J].Applied Catalysis A:General,2012,433-434:81-87.
[15]Han J S,Bredow T,DaveyD E,et al.The effect of Al addition on the gas sensing properties of Fe2O3-based sensors[J].Sens.Actuators B,2001,75:18-23.
[16]Qiu X,Miyauchi M,Sunada K,et al.Hybrid CuxO/TiO2nanocomposites as risk-reduction materials in indoor environments[J].ACS Nano.,2012,6:1609-1618.
猜你喜欢
车用发动机(2021年5期)2021-10-31
陶瓷学报(2020年6期)2021-01-26
中学生数理化·中考版(2018年11期)2019-01-31
教学考试(高考化学)(2018年5期)2018-12-06
赤峰学院学报·自然科学版(2018年7期)2018-08-11
小型内燃机与车辆技术(2016年4期)2016-10-21
淮南师范学院学报(2015年3期)2015-03-22
河北科技大学学报(2015年5期)2015-03-11
无机化学学报(2014年4期)2014-02-28
应用技术学报(2014年1期)2014-02-28
