
催化剂浓度对超支化聚4-氯甲基苯乙烯支化度的影响
2013-10-09 11:19周志平王晶晶贾正伟盛维琛
江苏大学学报(自然科学版) 2013年5期
周志平,王晶晶,贾正伟,盛维琛
(江苏大学材料科学与工程学院,江苏镇江212013)
支化度(DB)是表征超支化聚合物结构最重要的参数之一.近年来,超支化聚合物已经替代线形聚合物,被镶嵌到无机介孔材料,制备了具有大量末端官能团的有机-无机复合材料[1].末端官能团的数量主要是由超支化聚合物的支化度确定的,因此,提高和控制聚合物的支化度成为超支化聚合物研究领域中的热点[2].自缩合乙烯基聚合反应(SCVP)的出现,使得聚合物支化度比较容易得到控制[3-4].J.M.J.Fréchet等[3]以 3-(1- 氯乙基)苯乙烯为单体,通过阳离子聚合形成超支化聚苯乙烯.S.G.Gaynor等[5]以类似的引发单体4-氯甲基苯乙烯(4-CMS),通过过渡金属催化的原子转移自由基聚合(ATRP)也合成了超支化聚苯乙烯.M.W.Weimer等[6]通过1H和13C核磁共振(NMR)方法分析发现,催化剂浓度对聚合产物结构有很大影响.H.Komber和U.Georgi等[7-8]采用不同聚合方法及催化剂研究体系中各结构单元的影响,并计算出最终产物的支化度,但未系统研究催化剂浓度对支化度影响.
本研究以4-氯甲基苯乙烯为自引发单体,CuCl和2,2'-联吡啶(Bipy)为催化剂,通过ATRP溶液聚合方式,改变自引发单体和催化剂的摩尔比,合成不同支化度的超支化聚4-氯甲基苯乙烯[P(4-CMS)].利用1H和13C核磁共振,结合一维和二维核磁谱图对聚合物结构进行分析,确定其支化度.
1 试验
1.1 原料
4-氯甲基苯乙烯(质量分数 90%),美国Sigma-Aldrich公司;2,2'-联吡啶(质量分数99.5%),成都市科龙化工试剂厂;氯化亚铜(CuCl,质量分数97%),国药集团化学试剂有限公司;四氢呋喃(THF),成都市科龙化工试剂厂;无水乙醇,成都市科龙化工试剂厂.以上均为分析纯.氯苯,国药集团化学试剂有限公司,为化学纯.
1.2 超支化聚4-氯甲基苯乙烯的合成
图1为Cu(bipy)2Cl催化下,以4-氯甲基苯乙烯为自引发单体合成的超支化聚4-氯甲基苯乙烯.图1中,催化剂催化氯甲基形成苄基自由基,该自由基能引发双键反应,双键反应后成为新的活性中心,又能继续引发双键反应.聚合产物中有5种不同的结构单元,分别称为起始单元F、端点单元T、乙烯基型线性单元LV、缩合型线性单元LC和支化单元D.
在100 mL洁净干燥的三口烧瓶中加入4-氯甲基苯乙烯(1 g,5.897 mmol),2,2'-联吡啶(0.37 g,2.358 mmol),氯苯(20 mL);通氮气 5 ~10 min,再加入氯化亚铜(CuCl,0.12 g,1.179 mmol),常温搅拌5 min,使溶液均匀,成为深褐色;油浴锅中升至115℃,氮气保护下,反应适当时间.结束后,冷却至室温,加入适量四氢呋喃,空气中搅拌使未反应的亚铜离子充分氧化,形成络合物沉淀;静置数小时后,过滤,除去沉淀物;所得淡黄色聚合物溶液浓缩后,加入适量无水乙醇沉淀,沉淀物放入真空烘箱,45℃干燥.产品的产率用重量法测得.同样方法,改变催化剂浓度得到一系列聚合产物.

图1 聚合反应原理
1.3 表征
聚合物分子量通过凝胶渗透色谱仪(GPC)测得,GPC在Polymer Laboratories公司的PL-GPC 220上进行,THF为流动相,流速为1.0 mL·min-1,室温下测试.所有的核磁1H[400.13 MHz,δ(10-6)]和13C[100.62 MHz,δ(10-6)]分析都是在 Bruker AVANCEⅡ-400核磁共振仪上测得,采用5 mm BBO探头,温度为20℃.CDCl3作为溶剂时,以四甲基硅烷(TMS)为内标;以其他作为溶剂时,以溶剂峰作为内标.所用溶剂的溶剂峰为1H:δ=7.16(C6D6),2.05((CD3)2CO);13C:δ=77.16(CDCl3),128.06(C6D6).1H NMR 在1H 90°脉冲,采样时间3.98 s,延迟时间1 s时测得,定量的13C NMR采用反门控去偶测试方法,13C-1H HSQC(异核单量子)谱图通过Bruker提供标准脉冲序列测试记录.
2 结果与讨论
2.1 分子量与多分散性
最终的超支化聚4-氯甲基苯乙烯产物为淡黄色粉末状固体,易溶于苯、THF、丁酮等,不溶于水、乙醇等,产率约为85%,分子量通过GPC法测得,结果见表1.表中,Mn为聚合物数均分子量;Mw/Mn为多分散指数;x(T),x(LV),x(LC)和x(D)为各结构单元摩尔分数.x(A*)和x(B*)为活性中心自由基A*和B*摩尔分数.除1号外,Mn均为4 000左右,分子量分布较宽,Mw/Mn=5.47~8.32,与 M.W.Weimer等[6]研究结果相近.

表1 P(4-CMS)的表征结果
用GPC法测试分子量时,采用线形聚苯乙烯作为标样测得的超支化聚合物分子量比实际小.这是因为超支化聚合物具有三维准球形结构,其分子回转半径通常比相同分子量的线形聚合物小.据报道[6],采用GPC法测定的P(4-CMS)分子量都不高,一般为1 800~6 300.本研究主要讨论支化度,而超支化聚合物分子量的精确测定还有待改进.
2.2 聚合产物结构
各结构单元摩尔分数可通过核磁共振氢谱或碳谱进行计算.图2是 CDCl3为溶剂时3号样品1H NMR谱图.δ=1.5~3.2的峰是亚甲基和次甲基的质子相互重叠形成的;δ=6.0~7.5的峰是苯环上的质子峰;δ=5.2和5.7处是未反应的双键(═CH2)的质子峰,相应的双键(—CH═)的质子峰在δ=6.7处.

图2 聚合物样品3号的1 H NMR谱图
M.W.Weimer等[6]曾认为4.55与4.78处的2个峰分别是氯甲基(—CH2Cl)和氯亚甲基(—CHCl)上的质子峰.先对2个峰的13C NMR谱图进行分析,确定—CH2Cl和—CHCl上碳原子吸收峰.
图3为CDCl3作为溶剂,3号样品的13C NMR谱图.很明显13C NMR比1H NMR谱图的分辨率高.13C NMR谱图中各峰归属如下:δ=33是与苯环直接相连的亚甲基的碳原子吸收峰;δ=38.4处的一组宽峰是已反应的双键上亚甲基上碳原子吸收峰;δ=41.2和46.9是与氯亚甲基相连的亚甲基上碳原子吸收峰;δ=40.5和42.8处是次甲基吸收峰,其中高化学位移处的峰是与氯原子位置较近的次甲基碳原子吸收峰;δ=45.8和46.2处分别对应T和LV上氯甲基的碳原子吸收峰;δ=62.3和62.7处分别对应T和LC上氯亚甲基的碳原子吸收峰.苯环上碳原子吸收峰有2处:δ=125~130和135~145,高场的峰是未取代位置碳原子吸收峰.未反应双键上碳原子峰在113.3 (═CH2)和136.6(—CH═)处.

图3 聚合物样品3号的13 C NMR谱图
图4为 CDCl3作为溶剂时,3号样品的13C—1H HSQC谱图.从二维核磁谱图中,可清楚地看到氯亚甲基中碳原子吸收峰覆盖δ=4.3~5.0区域,氯甲基中碳原子吸收峰在δ=4.55处.LC上氯亚甲基的吸收峰在4.78处,而T上氯亚甲基吸收峰在δ=4.3~4.7,与氯甲基吸收峰 δ=4.55重叠.所以1H NMR谱图上4.78处对应的是LC上氯亚甲基吸收峰,而δ=4.55处是 T上CHCl和—CH2Cl重合峰.因此,在CDCl3溶剂中,无法区分开氯甲基和氯亚甲基的峰.但是,以 C6D6作为溶剂时,CHCl和CH2Cl在1H NMR谱图中质子峰可以很好地分开.
图5为C6D6作为溶剂,聚合物3号的1HNMR谱图.与CDCl3作为溶剂的1H NMR谱图相比,δ=4~5处的峰分开形成2组峰,归属如下:4.06和4.19处分别是T和LV上氯甲基的质子峰;4.55和4.66处分别是T和LC上氯亚甲基的质子峰.


以C6D6作为溶剂,13C NMR和13C—1H HSQC谱图如图6,7所示.


C6D6作为溶剂时,聚合物13C NMR谱图与CDCl3作溶剂时类似.从HSQC谱图上,可清楚看到CH2Cl和CHCl峰完全分开,这是由苯的溶剂化效应引起的.T和LV上氯甲基质子峰也得到很好地分开,从而可通过图中各区域积分面积确定聚合产物中各结构单元摩尔分数及支化度,结果见表1.
按照文献[7]计算方法,各结构单元积分值I(T),I(LV)和I(LC)分别通过图7的3个区域和I的积分面积来确定,即I(T)b'
I(LV)=Ib/2,I(LC)=的积分值为
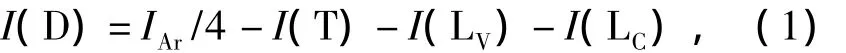
其中IAr为苯环上所有质子的积分,通过采用(CD3)2CO作为溶剂,比较CHCl/CH2Cl区域的积分面积来确定.
对于低分子量聚合物,IAr需减去未反应双键(—CH═)上质子的积分,双键上质子积分通过对(═CH2)积分并除以2得到.表1支化度表达式如下:

结果表明:催化剂和自引发单体的摩尔比为0.01时,P(4-CMS)的支化度很小,约为15.6%;随着催化剂用量的增大,P(4-CMS)支化度急剧增大,摩尔比为0.1和0.2时分别达到47.4%和48.6%;催化剂用量继续增大,支化度又呈下降趋势.
3 动力学分析
理论分析时[4,9],通常把 SCVP 反应单体称为AB型(A代表双键,B代表氯甲基),B被引发激活后成为活性中心B*(一级苄基自由基),双键A与B*反应后也成为活性中心A*(二级苄基自由基),A*也可以与A反应.因此,反应体系中同时存在2种活性中心自由基A*和B*,它们反应活性的差异,对最终产物结构也有很大影响,直接决定了反应体系中活性中心自由基摩尔分数.表1中活性中心自由基A*和B*的摩尔分数是根据文献[4]中的式(8b)和(8c)计算列出的.活性中心自由基摩尔比中,从1-8号是单调递增的.Yan Deyue等[4]对理想状态的SCVP反应推导出的活性中心反应速率常数比r为

式中:x为双键转化率;kA,kB分别为A*和B*的反应速率常数.除1和2号样r>1外,3~8号样r=0.46~0.68.式(3)是以B基团完全转化为B*为前提的,而1号和2号样中催化剂浓度少,不足以完全激活体系中的B基团,因此计算结果可靠性不大.而3~8号样中,催化剂浓度相对较大,使得体系中B基团被激活的几率大大提高,因此,3~8号样计算结果可靠性好.这样可根据3~8号样计算结果,确定在该聚合反应过程中生成的A*的反应活性要比B*小.根据这一结论就可以很好地解释催化剂浓度很低或较高时产物支化度低的原因:催化剂浓度很低时,反应初期生成的一级苄基自由基摩尔分数较低,并快速的反应生成二级苄基自由基,从而链增长反应主要发生在A*上;而当催化剂浓度较高时,体系中B*自由基摩尔分数较高,且反应活性较大,从而使得B*成为主要的链增长中心.
参照文献[10-11]中对非等速率自缩合乙烯基聚合反应的动力学分析,该反应体系中5种结构单元摩尔分数的解析表达式为

式中:xB为B*基团转化率.参数满足下列条件:
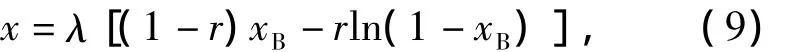
相应的平均支化度为

式(10)和(2)对支化度的定义略有不同,这里主要讨论反应结束的情况,此时两者一致.
以上动力学分析中,假定反应初期所有的催化剂都与自引发单体完全反应形成自由基,也就是说每个催化剂只能激活一个引发单体.而本合成试验中,催化剂激活存在一个平衡关系,即催化剂激活引发单体后并未消失,还能重新与链自由基钝化还原,再次激活引发单体.为此,提出一个催化剂平均激活次数m的概念,将式(10)改写为

式中:λ为催化剂与自引发单体的摩尔比.r=0.46 ~0.68,其平均值为0.55,x=1,当m=2.6 时,式(11)的DB与λ关系与试验结果一致(见图8).
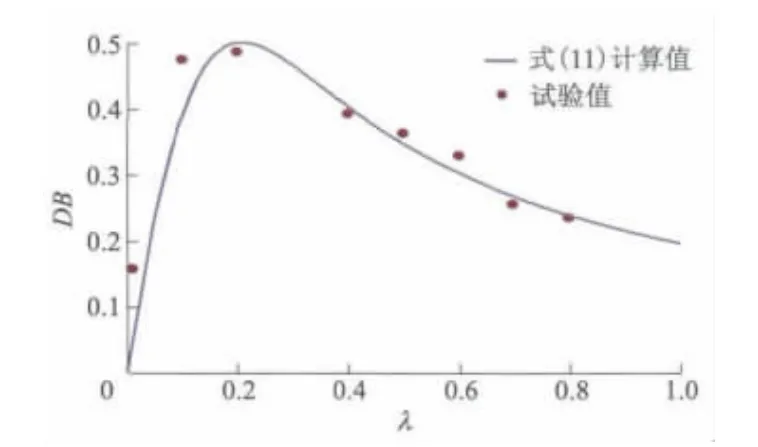
图8 支化度随催化剂浓度变化的关系曲线
4 结论
1)改变催化剂浓度是调控超支化聚4-氯甲基苯乙烯支化度的有效途径.当催化剂与自引发单体的摩尔比很低或较高时,生成的P(4-CMS)支化度均较低;当摩尔比在0.1~0.2之间时,P(4-CMS)支化度较高,接近于0.5.
2)计算结果表明:反应过程中生成的二级苄基自由基A*的反应活性要比一级苄基自由基B*的小.对照聚合反应的动力学分析表明:反应过程中活性中心A*和B*的反应速率常数比约为0.55,平均一个催化剂单体可以激活基团活化2.6次.
References)
[1] 周志平,张际亮,盛维琛,等.新型功能性介孔吸附材料的表征与吸附性能[J].江苏大学学报:自然科学版,2010,31(1):45-48.Zhou Zhiping,Zhang Jiliang,Sheng Weichen,et al.Characterization and adsorption property of new mesoporous adsorption material[J].Journal of Jiangsu University:Natural Science Edition,2010,31(1):45-48.(in Chinese)
[2] Radke W,Litvinenko G,Müller A H E.Effect of coreforming molecules on molecular weight distribution and degree of branching in the synthesis of hyperbranched polymers[J].Macromolecules,1998,31:239-248.
[3] Fréchet JM J,Henmi M,Gitsov I,et al.Self-condensing vinyl polymerization:an approach to dendritic materials[J].Science,1995,269:1080-1083.
[4] Yan Deyue,Müller A H E,Matyjaszewski K.Molecular parameters of hyperbranched polymers made by self-condensing vinyl polymerization,2:degree of branching[J].Macromolecules,1997,30:7024-7033.
[5] Gaynor SG,Edelman S,Matyhaszewski K,et al.Synthesis of branched and hyperbranched polystyrenes[J].Macromolecules,1996,29:1079-1081.
[6] Weimer M W,Fréchet JM J,Gitsov I.Importance of active-site reactivity and reaction conditions in the preparation of hyperbranched polymers by self-condensing vinyl polymerization:highly branched vs.linear poly[4-(chloromethyl)styrene]by met al-catalyzed ″living″radical polymerization[J].J Polym Sci,Part A:Polym Chem,1998,36:955-970.
[7] Komber H,Georgi U,Voit B.1H and13C NMR spectra of highly branched poly(4-chlormethylstyrene):signal assignment,structure characterization,and a SCVP kinetic study[J].Macromolecules,2009,42:8307-8315.
[8] Georgi U,Erber M,Stadermann J,et al.New approaches to hyperbranched poly(4-chloromethylstyrene)and introduction of various functional end groups by polymer-analogous reactions[J].Journal of Polymer Science,Part A:Polymer Chemistry,2010,48:2224-2235.
[9] 周志平,张际亮,盛维琛,等.非等速率自缩合乙烯基聚合反应产物的支化度[J].化学学报,2008,66(22):2547-2552.Zhou Zhiping,Zhang Jiliang,Sheng Weichen,et al.Degree of branching of products formed from general selfcondensing vinyl polymerization with non-equal reactivities[J].Acta Chimica Sinica,2008,66(22):2547-2552.(in Chinese)
[10] Zhou Zhiping,Wang Gaijuan,Yan Deyue.Kinetic analysis of self-condensing vinyl polymerization with unequal reactivities[J].Chinese Science Bulletin,2008,53(22):3516-3521.
[11] Zhou Zhiping,Yan Deyue.A general model for the kinetics of self-condensing vinyl polymerization[J].Macromolecules,2008,41:4429-4434.
猜你喜欢
油气·石油与天然气科学(2021年12期)2021-12-11
合成树脂及塑料(2020年4期)2020-09-20
中小学班主任(2019年12期)2019-09-10
山东化工(2019年13期)2019-02-16
分析化学(2017年12期)2017-12-25
新课程·中旬(2016年12期)2017-05-08
中学生数理化·高二版(2017年2期)2017-04-19
中国塑料(2015年5期)2015-10-14
粘接(2015年6期)2015-01-06
世界热带农业信息(2014年11期)2015-01-05
