吡啶掺杂碳载钴酞菁催化氧还原的电化学性能及在燃料电池中的应用
2013-09-17 06:59戴先逢郑明富石晶晶马承禺乔锦丽
物理化学学报 2013年8期
戴先逢 郑明富 徐 攀 石晶晶 马承禺 乔锦丽,*
(1东华大学环境科学与工程学院,上海201620;2浙江吉利控股集团有限公司,上海201501)
1 引言
近些年来,以碱性阴离子交换膜为传导介质的碱性阴离子交换膜燃料电池(AAEMs)由于其价格和性能上的优势引起世界范围研究者的极大兴趣与关注.同液体碱性燃料电池(AFCs)和质子交换膜燃料电池(PEMFC)相比,碱性阴离子交换膜燃料电池具有以下优点:(i)阳极和阴极具有更快的表面动力学,(ii)甲醇或乙醇燃料的渗透问题大大减小,(iii)阴极侧的水管理变得更为简单化,(iv)更宽的材料选择范围包括集流体、双极板,特别是在氧阴极可以使用非贵金属催化剂等.1-3其中以金属卟啉和酞菁为代表的M-N4大环化合物由于具有高的共轭结构和化学稳定性,11被认为是最有希望替代铂系金属的一类氧还原催化剂.然而无论从催化剂的稳定性还是活性而言,这些过渡金属大环化合物的性能仍然低于铂系金属,同时由于大环配体本身结构的复杂性,相对制备价格仍然偏高.为了提高过渡金属大环化合物的电催化性能,国内外研究者做了大量工作,发现制备高活性催化剂主要取决于以下几种关键因素:过渡金属源、12-19氮源20-24和碳载体,25为了提高催化剂在酸性介质中的稳定性还要进行热处理.8-11其中氮源对氧还原反应活性的提高起着至关重要的作用.
Collman26和Qiao27在研究烃类物质的氧化过程中发现用吡啶、咪唑及其衍生物等含氮小分子作为轴向配体在温和条件下即可活化分子氧,使烃类物质在空气作用下高效、高选择性地得以催化氧化.在我们的前期工作中,以吡啶(Py)小分子作为含氮配体,硫酸钴作为过渡金属源制备了高效氧还原碳负载钴吡啶催化剂(CoPy/C),发现Py可以协同金属Co显著提高CoPy/C催化氧还原的性能.26吡啶是一种结构简单、价格低廉的常规富氮源配体,然而,以吡啶分子作为含氮配体修饰金属大环化合物用于制备碳负载燃料电池氧阴极催化剂的研究却未见报道.基于此,本文以碳黑(Vulcan XC-72R)为载体,以Py作为直接氮源配体,与具有高共轭结构的钴酞菁协同作为催化剂前驱体,经溶剂分散法制备了碳负载吡啶氮掺杂的纳米钴酞菁复合催化剂(Py-CoPc/C);运用扫描电子显微镜-能谱仪(SEM-EDS)和X射线衍射技术(XRD)以及X射线光电子能谱(XPS)对催化剂的表面组成和微观结构进行了表征,并通过线性扫描伏安法(LSV)和旋转圆盘电极(RDE)技术等电化学手段对Py-CoPc/C在碱性介质中催化氧还原的性能、选择性以及催化机理进行了研究.同时将Py-CoPc/C催化剂用于制备膜电极组装(MEA)进行了H2-O2燃料电池发电性能的初步测试.
2 实验部分
2.1 实验仪器
扫描电子显微镜-能谱(SEM-EDS)分析利用JSM-5600LV型扫描电子显微镜进行,测定所用的加速电压为10 kV.D/Max-2550 PC型全自动X射线衍射仪(日本RIGAKU),Cu Kα辐射,波长0.154056 nm,管电压40 kV,管电流200 mA·cm-2,扫描速率15(°)·min-1,扫描角度为10°-90°.
X射线光电子能谱分析分析在ESCALAB250型高性能电子能谱仪进行测试,采用Al Kα辐射测定,功率250 W,光管电压为14 kV.
2.2 催化剂的制备
实验采用溶剂分散法制备吡啶掺杂钴酞菁复合催化剂.具体实验方法如下:以Vulcan XC-72R为碳载体,按照设定的比例(具体参见2.1节)将碳粉、吡啶和钴酞菁分散于20 mL甲醇(分析纯)溶液,然后将混合物置于玛瑙研钵研磨2 h,直至甲醇完全挥发.将混合物置于真空烘箱、40°C恒温2 h得到催化剂均一混合物粉末.为便于讨论,文中所有样品催化剂均按照原始催化剂合成所设定比例标记为x%Py-y%(CoPc)/C,其中x%和y%均为质量分数.
2.3 电极的制备
称取上述混合物粉末,分散于异丙醇(分析纯)中,异丙醇的用量与混合物粉末质量比均为1:2,超声10 min使之分散均匀,用移液枪移取2次5 μL混合均匀的催化剂浆液于玻碳电极(GC)表面,待自然晾干后再滴加一滴50:1(质量比)甲醇/Nafion溶液,自然晾干,玻碳电极表面上催化剂的载量为70.6 μg·cm-2.
2.4 电化学测试
电化学测试所用仪器为CHI760D电化学工作站(上海辰华公司).电化学测试采用三电极体系,以直径为0.5 mm的铂丝作为对电极,以饱和甘汞电极(SCE)作为参比电极,以玻碳电极(表面积为0.283 cm2)作为工作电极.上述制备的催化剂均在0.1 mol·L-1KOH电解质溶液中进行极化曲线测量,扫描速率分别为50和5 mV·s-1;实验开始前先通入N260 min,然后通入O230 min进行测试,实验均在室温下进行.循环伏安(CV)和旋转圆盘电极实验利用5908Triangle Drivec PINE电极(美国Raleigh公司).
2.5 H2-O2单电池测试
催化剂浆液以上述所制备的20%Py-20%CoPc/C与5%(w)Nafion溶液和异丙醇混合,超声4 h配制而成,其中催化剂与Nafion的质量比为3:1.将配制好的催化剂浆液喷涂到商业碳纸(Toray TGPH-090)上,使催化剂载量达到1.5 mg·cm-2作为阴极.同样方法制备得到阳极,所不同的是喷涂于碳纸上的为商业40%Pt/C催化剂(Johnson Matthey),Pt载量为0.5 mg·cm-2.为便于讨论,以未掺杂的40%CoPc/C在相同条件下同样制备阴极作为对比.然后将3滴阴离子粘结剂(Tokuyama AS-4,5%)涂覆于催化层表面,自然晾干.最后将阳极和阴极在70°C下热压于阴离子交换膜(Tokuyama A201膜,厚度28 μm,)的两面,制成膜电极(MEA),压力10 MPa,热压时间3 min,MEA有效活性面积为4 cm2.
将所制备的MEA进行H2-O2单电池测试,其中H2和O2的流量分别100和60 mL·min-1,室温下增湿.极化曲线在燃料电池测试平台系统GE/FC1-100上获得.
3 结果与讨论
3.1 电化学性能
催化剂的载量对氧还原反应的进程有着重要影响,27-29为了了解钴酞菁含量对催化氧还原性能的影响,首先以不同含量的CoPc负载于Vulcan XC-72R碳载体制备了不同载量的CoPc/C催化剂.图1是不同载量的CoPc/C催化剂于室温下O2饱和的0.1 mol·L-1KOH溶液中的ORR极化曲线.从图中可以看出,当钴酞菁载量从10%(10%CoPc/C)增加到40%(40%CoPc/C)时,ORR性能总体成增加趋势,并在钴酞菁载量达到40%时其催化性能达到最高.此时,40%CoPc/C的半波电位(E1/2)相对于10%CoPc/C正移了30 mV,并且在0.2-0.35 V电势范围内呈现出很好的扩散平台.然而,随着钴酞菁载量进一步增大到60%时(60%CoPc/C),ORR性能又迅速下降,起峰电位和半波电位分别比40%CoPc/C负移了45和90 mV,最大极限电流密度下降了46%.为了更加明确CoPc载量对催化氧还原性能的影响,图1插图中给出了在两个不同电势(-0.025和-0.15 V)下的ORR电流密度随CoPc载量的变化情况.由图1插图可以明显看出ORR电流密度随着CoPc载量的增加而逐渐增大,当CoPc载量大于40%时,ORR电流密度又呈下降趋势.这可能是由于当催化剂载量过高时,催化剂容易发生团聚现象,27-29比表面积下降从而使催化剂与氧接触的活性位减少,导致催化剂活性下降.

图1 不同CoPc含量的CoPc/C催化剂在室温下0.1 mol·L-1KOH电解液中氧饱和状态下的氧还原极化曲线Fig.1 Polarization curves for the ORR catalyzed by CoPc/C with different CoPc loadings,in O2-saturated 0.1mol·L-1KOH electrolyte at room temperature
基于上述结果,本实验将催化剂载量固定为40%,改变Py和CoPc的组成(以x%+y%=40%)进一步考察了Py和CoPc组成变化对氧还原性能的影响,如图2所示.为了便于讨论,40%Py/C在相同条件下的极化曲线也给出在图2中.从图2可以明显看出,当掺杂Py含量从10%增加到20%时,Py-CoPc/C催化氧还原的性能随Py含量的增加而增加.当Py含量为20%时,催化剂的活性达到最高,-0.2 V时电流密度提高到3.2 mA·cm-2,半波电位提高到-0.03 V.随着催化剂中Py含量的继续增加,电极的催化活性不再提高,而是随Py含量的增加而急剧下降.当Py的含量提高到35%,CoPc含量减小至5%时,Py-CoPc/C催化氧还原的起始电位负移了近140 mV,半波电位也由掺杂20%Py时的-0.03 V负移至-0.15 V.

图2 不同比例Py-CoPc/C的催化氧还原极化曲线Fig.2 Polarization curves for the ORR catalyzed by Py-CoPc/C with different ratios
根据文献,CoPc/C对O2的还原取决于O2分子在钴酞菁分子表面的吸附和脱附两个过程,其中O2的吸附是通过O2分子中的O原子与中心Co2+离子之间的配位作用实现.由于O原子与中心Co2+离子之间存在强的配位作用,致使反应产物和未反应的O2分子在催化剂表面的脱附变得困难,进而导致CoPc/C的氧还原活性降低.30当有吡啶掺杂时,吡啶N与钴酞菁之间形成具有轴向配位的Py-CoPc复合结构,30-32可以削弱O原子与中心Co2+离子之间强的配位作用,有利于活化分子氧,从而使催化剂的氧还原活性增强.此外,由于吡啶N位于CoPc分子的环外,可以和O2分子上的离域π电子以及Pc环上的离域π电子形成π-π作用,以提供更多O2吸附位,不仅易于氧分子在催化剂表面的吸附,同时易于产物和未反应的O2分子在催化剂表面的脱附.30然而,当进一步增大催化剂中Py的含量时,吡啶N原子上的孤对电子完全占据Co2+离子空的d轨道,促使活性中心Co2+离子无法与O2分子配位吸附,不利于活化分子氧.30同时,从另一个角度看,随着催化剂中Py含量的进一步增加,钴酞菁的含量随之减少.由于钴酞菁是含有八个N原子的大环化合物,CoPc含量的减少一方面使得氮源急剧下降,同时金属源也随之减少,两者共同作用的结果促使Py-CoPc/C催化氧还原的性能降低.值得一提的是,单纯的Py/C对ORR几乎不表现出催化活性(图2),然而,20%Py-20%CoPc/C对氧还原的催化活性与40%CoPc/C相比得到明显提高.相比于单纯的Py/C和CoPc/C催化剂,20%Py-20%CoPc/C催化剂的半波电位分别提高了160和15 mV.由此表明在CoPc/C催化剂上O2的吸附、还原产物的脱附以及电子的转移由于Py的掺杂而变得更加容易.
为了进一步明确Py掺杂及其含量对CoPc/C催化氧还原性能的影响,本文固定CoPc载量为20%,增加吡啶的含量在10%-40%范围,由图3给出了Py-CoPc/C室温下O2饱和的0.1 mol·L-1KOH溶液中的极化曲线.由图3(a)可以明显看出:当Py含量由10%增大到30%时,Py-CoPc/C催化氧还原的活性,无论是表现在半波电位还是极限扩散电流,均明显优于未掺杂的CoPc/C,并且当Py含量为20%时,催化剂的催化活性达到最高.然而,当Py含量继续增加到40%时,催化剂的催化活性不再提高,而是随Py含量的增加而下降.由图3(a)可以看出,当Py的含量提高到40%,总载量为60%时,半波电位负移至-0.04 V,同时极限扩散电流密度(-0.2 V)相比于掺杂20%吡啶含量时下降了42.9%,比未掺杂的CoPc/C下降了20%.这一现象与图1和图2中的结论完全一致,进一步说明了催化剂中过高的Py掺杂含量可能会削弱吡啶N与钴酞菁共轭结构的轴向配位效应,不利于活化分子氧,从而使Py-CoPc/C催化氧还原的性能又有所下降.
图3(b)是经过传质校正后不同Py含量的Py-CoPc/C催化氧还原反应的塔菲尔(Tafel)图,32,33以进一步考察Py掺杂对ORR动力学参数的影响.Tafel曲线由图3(a)的极化曲线获得,测试条件为O2饱和的0.1 mol·L-1KOH溶液,扫描速率为5 mV·s-1,电极转速为1500 r·min-1.由图3(b)可以看出,对于不同的电极,均表现为两个Tafel斜率,分别在低过电势区(E<-0.05 V)和高过电势区(E>0.00 V).这种Tafel斜率与电极电势的依赖关系可归因于“协同效应”和“抑制效应”,33-35其中不同Py含量掺杂的CoPc/C对ORR活性的差异性可以从低电流密度下的Tafel斜率明显判定出.对于未掺杂的CoPc/C,Tafel斜率为77.3 mV·dec-1,而对于10%Py-20%CoPc/C、20%Py-20%CoPc/C、30%Py-20%CoPc/C以及40%Py-20%CoPc/C,Tafel斜率分别为73.8、71.0、72.2和80.2 mV·dec-1,除40%Py-20%CoPc/C外均明显低于未掺杂的CoPc/C,其中20%Py-20%CoPc/C表现出最小的Tafel斜率,即71.0 mV·dec-1.以上结果充分说明ORR的反应机理与CoPc/C中Py的掺杂量密切相关,包括ORR过程中总的电子转移数的不同,这将在下面进一步讨论.

图3 (a)不同比例Py含量Py-CoPc/C(wCoPc=20%)催化剂在氧气饱和下0.1 mol·L-1KOH溶液中的氧还原极化曲线和(b)催化剂的塔菲尔图Fig.3 (a)Polarization curves for the ORR catalyzed by Py-CoPc/C(wCoPc=20%)with various Py loadings in O2-saturated 0.1 mol·L-1KOH electrolyte at room temperature and(b)its Tafel plots
O2的电化学还原是一个多电子过程,涉及的两个主要反应途径:经2e-转移生成H2O2和直接的4e-途径生成H2O.在碱性介质中,2e-途径可以表示为:

而直接的4e-途径可以表示为

为了获得最大的能量容量,希望O2的还原经历途径(2),即4e-过程.因而,为了进一步阐明CoPc/C在Py掺杂作用下的氧还原反应历程,本文运用RDE技术研究了具有最佳性能的20%Py-20%CoPc/C催化剂在不同转速下,O2饱和的0.1 mol·L-1KOH溶液中的电流-电势曲线,如图4(a)所示.从图中可以看出,随电极转速的增大,极限扩散电流随之而升高,同时不同转速下的电流-电势曲线在-0.1--0.4V范围内均呈现出很好的扩散电流平台,表明催化剂在电极表面的上分散均一.进一步通过Koutechy-Levich(K-L)方程可以计算出氧还原反应过程每个氧分子转移的电子总数,如图4(b)所示,具体计算过程如下:36

式中B为Levich斜率,ω为旋转圆盘电极的转速,n为每个氧分子转移的电子数,F为法拉第常数,S为电极表面积,CO2是O2在电解质溶液里的饱和浓度,DO2为O2的扩散系数,v是O2的粘滞阻力系数.
由图4(a)插图所示,根据Py-CoPc/C催化剂在-0.35-0.25 V电势下的曲线斜率计算出每个氧分子转移电子数均值为2.38.图4(b)比较了在电势为-0.35 V时CoPc/C和Py-CoPc/C两种催化剂的K-L曲线,根据K-L方程可以计算出在CoPc/C电极上每个氧分子转移的电子数为1.96.由此可以看出,尽管在CoPc/C和Py-CoPc/C两种催化剂所修饰的电极上ORR经历的都是2e-过程,然而在20%Py-20%CoPc/C电极上ORR的选择性大大提高.换句话讲,40%CoPc/C催化剂所修饰的电极上产生的大量H2O2在20%Py-20%CoPc/C催化剂所修饰的电极上被极大的抑制.由此说明,吡啶N的掺杂不仅可以提高碳载钴酞菁催化氧还原的活性,同时还可极大改善对氧还原的选择性.

图4 (a)室温下20%Py-20%CoPc/C催化剂在氧气饱和下0.1 mol·L-1KOH溶液中的RDE图及(b)不同催化剂在-0.35 V的电势下的Koutecky-Levich曲线图Fig.4 (a)RDE curves of 20%Py-20%CoPc/C catalyst in O2-saturated 0.1 mol·L-1KOH solution at room temperature and(b)Koutecky-Levich plots at the potential of-0.35 V on different catalysts
3.2 催化剂的组成和结构表征
3.2.1 EDS分析

图5 不同吡啶含量Py-CoPc/C催化剂中N的含量(w)Fig.5 N content(w)on the Py-CoPc/C with various Py loadings
为了进一步研究吡啶掺杂后碳载钴酞菁催化剂的组成,特别是N的存在状况,运用EDS获得的不同Py掺杂的CoPc/C中的氮含量如图5所示.EDS测试结果显示:CoPc/C催化剂中N元素的质量分数随着Py掺杂含量的增加而明显增加,当Py掺杂量为30%时(30%Py-20%CoPc/C),30%Py-20%CoPc/C催化剂中N含量达到13.9%,显然Py在催化剂中作为辅助氮源存在,并且和CoPc/C一起对氧还原进行协同催化.这与图2和图3中所得到的ORR测试结果完全相吻合.由此表明与单纯的CoPc/C和Py/C相比,通过吡啶N的掺杂修饰可能形成了具有轴向配位的高氮含量的Py-CoPc复合结构.如上述讨论可知,该Py-CoPc复合结构可以提供更多的O2吸附位,并同O2分子上离域π电子形成π-π作用,不仅易于O2分子在催化剂表面的吸附,同时易于产物和未反应的O2分子在催化剂表面的脱附,因而吡啶N修饰的碳载钴酞菁表现出更好的氧还原催化活性.32
3.2.2 XRD分析

图6Py-CoPc/C和CoPc/C的XRD图Fig.6 XRD patterns of Py-CoPc/C and CoPc/C catalysts
图6 是Py掺杂前后CoPc/C催化剂的XRD图谱,以进一步确定Py掺杂对CoPc/C催化剂的组成结构和催化活性位形成的影响.碳载体Vulcan XC-72R是典型的非晶态物质,在25°宽的衍射峰对应无定形碳的特征衍射峰.图中2θ在6°-7°处出现尖而强的衍射峰对应于CoPc的特征峰.37比较XRD结果可以看出,CoPc/C经Py掺杂N修饰后,其晶体结构发生了明显的变化,加入Py后2θ在6°-7°时的衍射峰强度明显减弱;此外,CoPc在2θ为26.3°和27.5°出现的强衍射峰在Py掺杂后消失.由此表明由于Py的加入,使CoPc由晶形态进一步向无定形态转变,使催化剂比表面积增大,从而可以提供更多的O2吸附位,有利于催化剂氧还原反应的进行,这与上述所观察到的电化学测试结果与EDS分析结果一致.
3.2.3 XPS分析
考虑到EDS特别是对原子量较小元素测试结果存在偏差性,本文对Py-CoPc/C催化剂进行了XPS能谱测试,并采用同样方法制备了PyCo/C和CoPc/C催化剂作为对照,以进一步明确Py掺杂前后催化剂表面组成中氮元素有不同形式的化学键合态.采用高斯法对N 1s的XPS图谱进行分峰拟合,结果如图7所示.由图7(a)可以看出,对PyCo/C中N 1s分峰拟合得到2条重复性较高的曲线,结合能分别位于400.1和401.5 eV,可分别归属为吡咯N/吡咯酮N和季铵式N(石墨N);对CoPc/C中N 1s分峰拟合同样得到2条重复性较高的曲线(图7(b)),结合能分别位于398.8和400.3 eV,分别归属为吡啶式N和吡咯N/吡咯酮式N.位于(400.0±0.3)eV的氮原子贡献出两个p电子与碳原子形成共价键,俗称为吡咯式的氮形式,而位于(398.7±0.2)eV结合能处的氮原子仅贡献一个p电子参与催化剂的成键,故结合能最低,称为吡啶式的氮形式.20,38,39由图7(c)发现对于20%Py-20%CoPc/C催化剂N 1s分峰拟合得到了3条重复性较高的曲线,其结合能分别位于398.8、399.3和401.2 eV,亦即经Py掺杂N修饰后,CoPc/C催化剂中同时存在有吡啶式N和石墨式N(季铵N)结构.大量的文献表明,吡啶式N和石墨式N对催化剂的催化活性起重要作用,而吡咯式的氮对氧还原没有催化活性.26,40,41这就是为什么观察到20%Py-20%CoPc/C催化剂的催化活性相比于单纯的Py/C和CoPc/C催化剂明显提高的原因(见图2和图3).结合LSV结果,可以推断吡啶式N和石墨式N配位于Co离子作为催化剂的活性中心,以提供氧还原活性位.由此,进一步表明Py掺杂可以作为辅助氮源存在,和CoPc/C一起对氧还原进行协同催化.
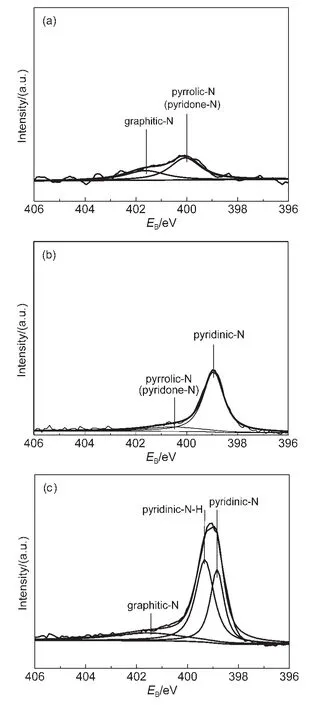
图7 PyCo/C(a),CoPc/C(b)和Py-CoPc/C(c)催化剂XPS N 1s图Fig.7 XPS N1 s spectra of PyCo/C(a),CoPc/C(b)and Py-CoPc/C(c)catalysts
3.3 催化剂的稳定性
催化剂的稳定性在O2饱和的0.1 mol·L-1KOH溶液中,在设定的-0.3-0.6 V电势范围内连续进行3000次循环伏扫描安测试,结果如图8所示.从图中可以看出,20%Py-20%CoPc/C催化剂总体表现出较好的稳定性,在最初的500次连续电势循环扫描中,无论是半波电位还是峰电流密度均无任何变化;经过3000次连续电势循环扫描后,起峰电位和半波电位基本上没有明显变化,只是电流密度有所下降,由最初的1.75 mA·cm-2下降到1.0 mA·cm-2.这可能是由于所制备的催化剂中碳的含量比较高,碳层包裹钴的大部分,从而能有效保护钴免受中间产物或其它杂质的侵袭,从而提高催化剂的氧还原性能和稳定性.

图8 室温下20%Py-20%CoPc/C催化剂在氧饱和0.1 mol·L-1KOH溶液中运行不同圈数后的ORR的CV图Fig.8 CV curves for the ORR with 20%Py-20%CoPc/C catalyst after different cycles in O2-saturated 0.1 mol·L-1 KOH at room temperature
3.4 H2-O2单电池性能
图9是以Tokuyama AS-4阴离子交换膜和上述所制备的催化剂制备的MEA在H2-O2燃料电池单电池中的发电性能.作为对比,以未掺杂的40%CoPc/C作为阴极催化剂,同样条件下制备了MEA.由图9可以看出,以单纯的CoPc/C所制备的MEA,获得的最大发电功率密度为8.6 mW·cm-2.当催化剂为Py-CoPc/C时,获得最大发电功率密度为21 mW·cm-2,是前者的2.4倍,同时开路电压(OCV)也由CoPc/C的0.61 V提高到Py-CoPc/C的0.86 V,最大发电电流密度由70 mA·cm-2提高到80 mA·cm-2.这里值得一提的是,在MEA制备过程中均没有进行最优化制备.这些初步结果进一步说明Py掺杂可以极大地提高CoPc/C的催化活性,减小活化极化,从而有望作为一种新型的碱性燃料电池氧阴极催化剂.

图9 H2-O2阴离子交换膜燃料电池发电性能Fig.9 Preliminary performance of a H2-O2anion exchange membrane fuel cell
4 结论
通过溶剂分散方法成功制备了碳负载吡啶氮掺杂钴酞菁复合催化剂(Py-CoPc/C).该类催化剂在碱性介质中对氧还原表现出良好的电催化活性,并且同Py掺杂的含量密切相关.当掺杂Py中氮含量为20%时,Py-CoPc/C催化氧还原的活性最高,在0.1 mol·L-1KOH电解质溶液中起峰电位达到0.24 V,半波电位为0.01 V,同时表现出有很好的扩散电流.RDE技术研究表明:Py的掺杂不仅可以提高CoPc/C催化氧还原的活性,同时还极大改善了对氧还原的选择性.与单纯40%Py/C和未掺杂的40%CoPc/C催化剂相比,Py-CoPc/C催化氧还原的半波电位分别提高了160和15 mV,O2分子在电极上反应的总电子转移数也由CoPc/C的1.96提高到Py-CoPc/C的2.38.EDS分析结果显示,催化剂中N元素含量随Py含量的增加而增加,表明Py在催化剂中作为辅助氮源存在,和CoPc/C一起对氧还原进行协同催化.XRD研究结果显示经Py氮掺杂后,CoPc/C催化剂由晶形态向无定形态转变,催化剂的分散更加均一,比表面积有所增大,从而更加有利于O2分子吸附.XPS分析表明吡啶N和石墨N结构均存在于Py-CoPc/C催化剂中,配位于中心金属离子Co对催化氧还原的活性发挥重要作用.CV稳定性测试结果表明Py-CoPc/C催化剂具有较好的稳定性.以Py-CoPc/C做氧阴极催化剂,H2-O2燃料电池单电池室温下获得最大发电功率密度21 mW·cm-2,是CoPc/C的2.4倍,表明Py-CoPc/C有望作为一种新型的燃料电池阴极催化剂.
(1)Abdel,R.M.A.;Abdel,H.R.M.;Khalia,M.W.J.Power Sources 2004,134,160.doi:10.1016/j.jpowsour.2004.02.034
(2) Tripkovic,A.V.;Popovic,K.D.;Grgur,B.N.;Blizanac,B.;Ross,P.N.;Markovic,N.M.Electrochim.Acta 2002,47,3707.doi:10.1016/S0013-4686(02)00340-7
(3)Chen,R.R.;Li,H.;Chu,D.;Wang,G.F.J.Phys.Chem.C 2009,113,20689.doi:10.1021/jp906408y
(4) Lefèvre,M.;Dodelet,J.P.Electrochim.Acta 2003,48,2749.
(5) Baker,R.;Wilkinson,D.P.;Zhang,J.Electrochim.Acta 2008,53,6906.doi:10.1016/j.electacta.2008.01.055
(6) Li,Z.P.;Liu,B.H.J.Appl.Electrochem.2012,40,475
(7) Yu,E.H.;Cheng,S.;Logan,B.F.;Scott,K.J.J.Appl.Electrochem.2009,39,705.doi:10.1007/s10800-008-9712-2
(8) Cote,R.;Lalande,G.T.;Bailey,L.D.;Guay,D.;Dodelet,J.P.J.Electrochem.Soc.1998,145,2411.doi:10.1149/1.1838651
(9) Lu,Y.;Reddy,R.G.Int.J.Hydrog.Energy 2008,33,3930.doi:10.1016/j.ijhydene.2007.12.031
(10) Bambagioni,V.;Bianchini,C.;Filippi,J.;Lavacchi,A.;Oberhauser,W.;Marchionni,A.J.Power Sources 2011,196,2519.doi:10.1016/j.jpowsour.2010.11.030
(11) Chung,H.T.;Johnston,C.M.;Zelenay,P.Electrochemical Society Trans.2009,25,485
(12) Faubert,G.;Cote,R.;Dodelet,J.P.;Lefevre,M.;Bertrand,P.Electrochim.Acta 1999,44,2589.
(13) Cote,R.;Lalande,G.;Faubert,G.;Guay,D.;Dodelet,J.P.;Denes,G.J.New Mater.Electrochem.Syst.1988,1,7.
(14) Gupta,S.;Tryk,D.;Zecevic,S.K.;Aldred,W.;Guo,D.;Savinell,R.J.Appl.Electrochem.1998,28,673.doi:10.1023/A:1003288609411
(15) Bhugun,I.;Fred,C.Anson.J.Electroanal.Chem.1997,430,155.
(16) Gouerec,P.;Savy,M.Electrochim.Acta 1999,44,2653.
(17) Seeliger,W.;Hamnett,A.Electrochim.Acta 1992,37,763.doi:10.1016/0013-4686(92)80083-X
(18)Tian,J.H.;Wang,F.B.;Shan,Z.Q.;Wang,R.J.;Zhang,J.Y.J.Appl.Electrochem.2004,34,461.doi:10.1023/B:JACH.0000021860.94340.02
(19) Cheng,H.;Yan,W.;Scott,K.Fuel Cells 2007,7,16.
(20) Nallathamibi,V.;Lee J.W.;Kumaraguru,S.P.;Wu,G.;Popov,B.N.J.Power Sources 2008,183,34.doi:10.1016/j.jpowsour.2008.05.020
(21) Ozaki,J.;Kimura,N.;Anahara,T.;Oya,A.Carbon 2007,45,1847.doi:10.1016/j.carbon.2007.04.031
(22) Iwazaki,T.;Obinata,R.;Sugimoto,W.;Takasu,Y.Electrochem.Commun.2009,11,376.doi:10.1016/j.elecom.2008.11.045
(23) Iwazaki,T.;Yang,H.;Obinata,R.;Sugimoto,W.;Takasu,Y.J.Power Sources 2010,195,5840.doi:10.1016/j.jpowsour.2009.12.135
(24) Gong,K.;Du,F.;Xia,Z.;Durstock,M.;Dai,L.Science 2009,323,760.doi:10.1126/science.1168049
(25) Lefèvre,M.;Proietti,E.;Jaouen,F.;Dodelet,J.P.Science 2009,324,71.doi:10.1126/science.1170051
(26) Collman,J.P.;Chien,A.S.;Eberspacher,T.A.;Zhong,M.;Brauman,J.I.Inorg.Chem.2000,39,4625.doi:10.1021/ic000071z
(27) Qiao,J.L.;Xu,L.;Ding,L.;Zhang,L.;Baker,R.;Dai,X.F.;Zhang,J.J.Appl.Catal.B 2012,125,197.doi:10.1016/j.apcatb.2012.05.050
(28)Velázquez-Palenzuela,A.;Zhang,L.;Wang,L.C.;Cabo,P.L.;Brillas,E.;Tsaya,K.;Zhang,J.J.Electrochim.Acta 2011,56,4744.doi:10.1016/j.electacta.2011.03.059
(29) Ding,L.;Qiao,J.L.;Dai,X.F.;Zhang,J.J.;Tian,B.L.Int.J.Hydrog.Energy 2012,37,14103.doi:10.1016/j.ijhydene.2012.07.046
(30)Xu,Z.W.;Li,H.J.;Cao,G.X.;Zhang,Q.L.;Li,K.Z.;Zhao,X.N.J.Mol.Catal.A-Chem.2011,335,89.doi:10.1016/j.molcata.2010.11.018
(31) Iamamoto,Y.;Assis,M.D.;Ciuffi,K.J.;Sacco,H.C.;Iwamoto,L.;Melo,A.J.B.;Nascimento,O.R.;Prado,M.C.J.Mol.Catal.A-Chem.1996,109,189.doi:10.1016/1381-1169(96)00030-1
(32) Baranton,S.;Coutanceau,C.;Roux,C.;Hahn,F.;Leger,J.M.J.Electroanal.Chem.2005,577,223.
(33) Lima,F.H.B.;Giz,M.J.;Ticianelli,E.A.J.Braz.Chem.Soc.2005,16,328.
(34) Giacomini,M.T.;Ticianelli,E.A.;McBreen,J.;Balasubramanianb,M.J.Electrochem.Soc.2001,148,A323.
(35) Markovic,N.M.;Schmidt,T.J.;Stamenkovic,V.;Ross,P.N.Fuel Cells 2001,1,105.
(36) Bard,A.J.;Faulkner,L.Electrochemical Methods,Wiley&Sons:New York,2001,p 331.
(37)Lu,Y.H.;Ramana,G.R.Electrochim.Acta 2007,52,2562.
(38) Li,X.;Liu,G.;Popov,B.N.J.Power Sources 2010,195,6373.(39) Lee,K.;Zhang,L.;Lui,H.Electrochim.Acta 2009,54,4704.
(40)Velázquez-Palenzuela,A.;Zhang,L.;Wang,L.J.Phys.Chem.C 2011,115,12929.
(41) Liu,G.;Li,X.;Ganesan,P.Electrochim.Acta 2010,55,2853.
猜你喜欢
潍坊学院学报(2021年2期)2021-07-22
昆明医科大学学报(2020年11期)2020-12-28
中西医结合肝病杂志(2020年2期)2020-10-27
化工学报(2020年4期)2020-05-28
今日农业(2019年11期)2019-08-13
安徽化工(2018年4期)2018-09-03
现代检验医学杂志(2016年1期)2016-11-12
浙江理工大学学报(自然科学版)(2015年7期)2015-03-01
现代检验医学杂志(2015年4期)2015-02-06
应用化工(2014年10期)2014-08-16