
4-碘吡唑铜配合物的合成、结构及量子化学
2013-09-15 03:03侯亚男魏东明邢永恒
无机化学学报 2013年10期
宋 鸽 孙 巧 侯亚男 张 瑞 魏东明 施 展 邢永恒*,
(1辽宁师范大学化学化工学院,大连 116029)
(2澳大利亚昆士兰大学生物工程及纳米科技学院计算分子科学研究中心,布里斯班 4072,澳大利亚)
(3无机合成与制备化学国家重点实验室,吉林大学化学学院,长春 130012)
在配位化学中,吡唑类配体一直都是人们研究的热点之一,而吡唑类化合物是一类结构简单并且非常重要的杂环化合物,有着较好的生物活性及配位能力,并且在农业、医药、染料以及配位化学等领域都有广泛的应用[1-10]。以单吡唑作为有机配体与金属离子配位时主要存在两种配位形式:质子化和去质子化。当吡唑以质子化形式参与配位时,形成的配合物中通常存在分子内或分子间氢键作用,如:[ZnCl(HpzPh)3]Cl[11]和 trans-[PdCl2(Hpz)2][12]等;当以去质子化方式参与配位时,单吡唑起桥基的作用,即借吡唑的双齿桥联作用构筑环多核配合物如:[Cu3(μ3-OH)-(μ-pz)3(MeCOO)(MeO)(Hpz)][13],[Cu3(μ3-OH)(μ-pz)3(MeCOO)2(Hpz)][14]等。
近年来报道的一些铜吡唑配合物如:[3,5-(CF3)2Pz]Cu3[15]和[Cu3(μ3-OH)(μ-pz)3(py)3](CF3SO3)2·0.5H2O[16],其中金属铜原子的化合价分别是+1价和+2价,吡唑都作为桥配体参与配位。通过本课题组的大量实验研究发现:吡唑的4-位被碘取代后,其溶解性、配位能力、稳定性均发生改变,而且关于以4-碘吡唑为配体合成配合物的报道还很少。为了进一步系统的研究4-碘吡唑配合物在合成,性质及结构上的特征,因此我们首次成功的设计、合成了2个以4-碘吡唑为配体的配合物[Cu3(Ipz)3](1)和[Cu(SO4)(Ipz)4]·2H2O·CH3OH(2),它们是由共价键形成的环状和一维链状结构。
1 实验部分
1.1 仪器和试剂
JASCO FT/IR-480型傅里叶变换红外光谱仪(日本 JASCO公司,KBr压片, 波数为 200~4 000 cm-1);V-570型紫外光谱仪(日本 JASCO,波长范围200~2500 nm);Bruker SMART APEX Ⅱ CCD 型 X射线单晶衍射仪;PE 2400型CHN元素分析仪 (美国PE公司);日立F-7000荧光光谱仪(日本日立公司);实验所需药品 Cu(Ac)2·H2O、CuSO4·5H2O、4-碘吡唑、乙醇和甲醇均为分析纯试剂。
1.2 配合物的合成
1.2.1 配合物[Cu3(Ipz)3]的合成
将 Cu(Ac)2·H2O(0.105 2 g,0.5 mmol) 和 4-碘吡唑 (0.097 1 g,0.5 mmol)溶于 15 mL 无水乙醇中,室温搅拌0.5 h后用 KOH溶液调pH值为5~6,将混合液装入具有聚四氟乙烯内衬的不锈钢反应釜中,于120℃烘箱内静置加热5 d。取出冷却至室温,产物经过过滤和乙醇洗涤,室温干燥后,得到白色块状晶体。产率:52%(以铜为基准计算)。元素分析 (C9H6N6I3Cu3) 测定值 (%):C,14.03;H,0.82;N,10.89;理论值(%):C,14.05;H,0.78;N,10.92。
1.2.2 配合物[Cu(SO4)(Ipz)4]·2H2O·CH3OH 的合成
将 CuSO4·5H2O(0.105 2 g,0.5 mmol)和 4-碘吡唑(0.097 1 g,0.2 mmol)溶于 10 mL 无水甲醇中,室温搅拌2 h后过滤,滤液静置在自然条件下挥发,3 d后瓶底出现蓝色晶体,产物经过滤和甲醇洗涤,室温干燥后,得到蓝色块状晶体。产率:59%(以铜为基准计算)。元素分析(C9H6N6I3Cu3)测定值(%):C,15.49;H,2.09;N,11.13;理论值 (%):C,15.56;H,2.00;N,11.16。
1.3 X-ray单晶结构测定
选择大小为 0.38 mm×0.08 mm×0.06 mm 配合物 1 和 0.56 mm×0.49 mm×0.21 mm 的配合物 2 的晶体,在Bruker Smart APEXⅡCCD型X射线单晶衍射仪上,于293 K下使用经石墨单色化的Mo Kα光源(λ=0.071 073 nm),以 φ-ω 扫描方式收集衍射数据。配合物1共收集到6 505个衍射点,其中独立衍射点1 468个,强点1 310个(I>2σ(I))用于结构解析。配合物2共收集到7 149个衍射点,其中独立衍射点 5 041个,强点 3 318个(I>2σ(I))用于结构解析。衍射强度数据经Lp因子校正。晶体结构由直接法解出,对所有非氢原子坐标和各向异性温度因子进行全矩阵,最小二乘法修正。氢原子坐标由理论加氢程序确定。所有计算均用SHELX-97程序[17]在PentiumバPC计算机上进行。配合物的晶体学参数列于表1,配合物的分子结构及分子间弱的相互作用在SHELX-97程序和Diamond 3.2程序中画出。
CCDC:900585,1;900586,2。
2 结果与讨论
2.1 红外光谱
配合物1的红外光谱:峰位在3 111 cm-1处的强吸收峰归属为4-碘吡唑分子的=C-H的伸缩振动峰;1 629、1 285、1 054 cm-1处吸收峰归属为 4-碘吡唑中吡唑环的特征伸缩振动峰;与配体4-碘吡唑中吡唑环上的伸缩振动峰1 624、1 266、1 028 cm-1相比,配合物1的红外谱图中的相应峰位发生了蓝移。
配合物2的红外光谱:峰位在3 124 cm-1处的强吸收峰归属为4-碘吡唑环上的=C-H的伸缩振动峰;2 923和2 851 cm-1吸收峰归属为游离甲醇分子中甲基上的C-H伸缩振动峰;1614、1 285、1 055 cm-1吸收峰归属为吡唑环的特征伸缩振动峰与配体 4-碘吡唑环上 C=C、C-N伸缩振动峰 1 624、1 256、1 028 cm-1相比峰位分别发生了红移和蓝移;1 148和608 cm-1处的吸收峰归属为硫酸根的伸缩振动峰;峰位在3 423、1 557、1 508 cm-1的吸收峰可归属为配合物中游离水的特征峰。
2.2 紫外光谱
在配合物1的UV-Vis吸收光谱中,出现3个主要的吸收峰,分别在λmax=206、246和 398 nm。其中,λmax=206、246 nm处的吸收峰是配体自身的π→π*跃迁;λmax=398 nm的吸收峰归属为配体和中心金属离子间的荷迁移跃迁带(LMCT)。
在配合物2的 UV-Vis吸收光谱中,200~750 nm间出现4个主要的吸收峰,分别在λmax=208、262、346 和 642 nm。其中,λmax=208、262 nm 处的吸收峰是配体自身的π→π*跃迁;λmax=346 nm的吸收峰归属为配体和中心金属离子间的荷迁移跃迁带 (LMCT);λmax=642 nm的吸收峰归属为 Cu(Ⅱ)的d→d* 跃迁(2Eg←2T2g)[18]。
2.3 晶体结构
配合物1的X射线晶体分析表明,其晶体属于正交晶系,Pnma空间群。配合物1的主要键长、键角数据分别列于表2,在其不对称单元中包括半个晶体学独立的[Cu3(Ipz)3]分子。从图1可以看出Cuガ原子的配位环境相同,每个Cu(I)原子与来自2个4-碘吡唑配体的2个N原子 (Cu1与N1,N1i;Cu2与N2,N3)配位,形成平面三角形结构。在配合物的结构中,N-Cu-N 的键角范围是 173.0(2)°~178.6(1)°;Cu-N 的键长范围为 0.185 8(3)~0.187 1(3)nm,Cu1…Cu2 之间的距离是 0.333 64(3)nm,Cu2…Cu2i之间的距离是0.313 33(4)nm。与相应文献[19]相比,N-Cu-N 的键角在 174.5(3)°~178.2(3)°范围之内、Cu-N的键长 0.184 5(8)~0.187 6(7)nm 和 Cu1…Cu2 之间的距离 0.310 0~0.348 2 nm 都十分接近。 4-碘吡唑以去质子化形式作为桥联配体连接2个铜原子,形成一个Cu3N6九元环结构。

表2 配合物的主要键长和键角Table 2 Selected bond lengths(nm)and angles()of complex

图1 配合物1的分子结构图Fig.1 Structure motif of complex 1

图2 配合物2的不对称单元图Fig.2 Coordinate environment of Cu(Ⅱ) in complex 2
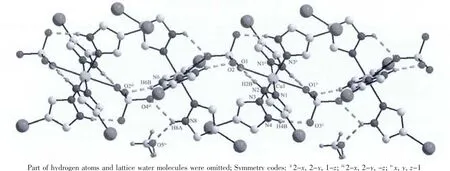
图3 配合物2的一维无限链状氢键结构Fig.3 Infinite 1D chain hydrogen bonding structure of complex 2

表3 配合物2的氢键Table 3 Hydrogen bonds of complex 2
配合物2的X射线晶体分析表明,其晶体属于三斜晶系,P1空间群。配合物2的主要键长、键角数据分别列于表2。如图2所示,每个不对称结构单元中有2个六配位的Cu(Ⅱ)原子,4个4-碘吡唑配体,一个配位的硫酸根离子,2个游离的水分子和1个游离甲醇分子。如图3所示,中心金属铜Cu1与4个配体上的4个氮原子 (N1、N3、N1ii和N3ii)和2个硫酸根分子中的2个氧原子(O1、O1ii)形成CuN4O2的扭曲八面体构型,Cu2的配位模式与Cu1相同。4-碘吡唑配体以质子化形式作为单齿配体参与配位,硫酸根离子作为桥配体通过共价键连接2个铜原子形成一条无限的一维链状结构(图3)。配合物2中的Cu-N键长有4种,分别为 Cu1-N1 0.202 4(8)nm,Cu1-N3 0.202 9(8)nm,Cu2-N5 0.199 6(9)nm和Cu2-N7 0.203 1(9)nm。N-Cu-N的键角范围是89.12(4)°~90.88(5)°,并且 Cu1…Cu2 之间的距离是0.644(6)nm。此外,在配合物的结构中存在大量的N-H…O(N2-H2B…O1,N2-H2B…O2,N4-H4B…O3ii,N6-H6B…O2iii,N8-H8A…O5iv和 N8-H8A…O4iii)分子间氢键作用,进一步加强了配合物结构的稳定性。详细的氢键键长、键角列于表3。
2.4 荧光光谱
在室温下,激发波长为 360 nm(狭缝 2.5∶2.5)的条件下,测定了固态配合物1的发射光谱。如图4,配合物1在418 nm处有荧光发射峰,归属为金属到配体的荷迁移(MLCT),这表明配合物1具有荧光特性,是潜在的发光材料。与相应文献[20]相比,配合物1的荧光谱带发生了一定程度的蓝移。这是由于配体向金属发生了荷迁移(MLCT)跃迁,配位原子的孤对电子参与大π键共轭,配体的电子分布改变,引起的配合物体系激发态能级升高所致。

图4 配合物1的荧光光谱Fig.4 Emission spectra of the complex 1
2.5 量化计算
运用Gaussian09量子化学程序包[21],采用密度泛函理论(DFT)[22]中的B3LYP方法[23]对配合物1和2的分子结构参数进行自然键轨道理论计算[24-25]。其中 C、H、N、S 和 O 原子采用 6-31G(d)基组,铜和碘原子选用赝势基组LANL2DZ[26]。计算中使用的分子构型参数均取自晶体结构实验数据 (没有考虑水和甲醇溶剂分子)。化合物1和2的HOMO和LUMO前沿轨道见图5和6。

图5 配合物1的前沿轨道图Fig.5 HOMOand LUMO of complex 1

图6 配合物2的前沿轨道图Fig.6 HOMOand LUMO of complex 2
2.5.1 稳定性分析
根据分子轨道理论,前线轨道和邻近分子轨道对于配合物的稳定性起着重要作用。最高占据与最低空轨道之间的能量差距越大,配合物的动力学稳定性越强。如表2所示:对于配合物1和2,它们的最高占据分子轨道(HOMO)的能量分别为-0.231 99和-0.197 25 a.u.,而最低的空轨道(LUMO)的能量分别为-0.046 68和-0.185 58 a.u.,2个轨道之间的能量差ΔE分别为(ELUMO-EHOMO)5.04和0.32 eV。结果分析表明配合物1的动力学稳定性强于2,这是由于它们的配位构型和配位环境不同所导致的。
从图5和6可以看出,配合物1的HOMO的电子云大都分布在4-碘吡唑配体上,LUMO的电子云大都分布在金属配位中心上,说明配合物1的紫外光谱中存在配体到金属的LMCT跃迁;对于配合物2的HOMO和LUMO的电子云均分布在金属配位中心和配位N和O原子上,说明配合物2的紫外光谱中存在配体到金属的LMCT跃迁和Cu(Ⅱ)d→d*跃迁。
2.5.2 电荷分析
配合物的原子净电荷计算结果显示,配合1的中心金属Cu1和Cu2原子上的净电荷分别是0.406 e和0.40 e,价层电子排列方式分别是4s0.513d9.764p0.12和4s0.523d9.764p0.13,这说明金属铜原子的d轨道参与成键。由于铜原子的化合价是+1价,而实际净电荷不到+1价,因此铜原子所获得的电子来源于4-碘吡唑配体。对于配位原子N(N1,N2和N3)的平均净电荷是-0.343 e。类似地,配合物2中Cu1和Cu2原子上的净电荷分别是0.537 e和0.530 e,价层电子排列方式是 4s0.283d9.464p0.465p0.01和 4s0.313d9.604p0.34,同样也是中心金属铜原子的d轨道参与成键。由于铜原子的化合价是+2价,而实际净电荷不到+2价,因此铜原子所获得的电子来源于4-碘吡唑配体和配位的硫酸根。对于配位原子N(N1,N3,N5和N7)的净电荷变化范围是从-0.368 e到-0.446 e,而O(O1和O4)的净电荷分别是-0.614 e和-0.604 e。通过比较两个配合物的净电荷可以发现:配合物1和2中铜原子的净电荷分别小于+1和+2价,说明金属铜从配体得到部分反馈电子,铜与配体形成了比较明显的共价键,金属与配体之间发生了荷迁移,进一步证实了配合物的紫外光谱性质。配合物2的净电荷数值比配合物1大,配位N原子上的净电荷比配合物1更负,这归咎为在形成配合物时配体的给电子能力不同。
Wiberg键级分析表明,在配合物1和2中,Cu-N键级分别是0.344 87和0.276 95,前者键级大于后者,表明前者的成键作用大于后者;配合物2中,Cu原子和N原子配位键相互作用较强,Cu原子和O原子配位键相互作用较弱,这与结构分析结果一致。
3 结 论
以4-碘吡唑为配体,采用2种不同的合成方法,首次成功合成了2种不同的4-碘吡唑铜配合物。其中配合物1是一个封闭的Cu3N6九元环状结构,而配合物2是以构造块“SO42-”作为桥连接“Cu(IPz)4”形成一维无限链状结构。本文对配合物结构、光谱特性及量化数据进行了详细的讨论。为进一步研究此类配合物的结构及功能特性提供了有价值的参考。
[1]Patel M V,Bell R,Majest S,et al.Org.Chem.,2004,69:7058-7065
[2]Morimoto KM,Makino K,Yamamoto S,et al.Adv.Heterocycl.Chem.,1990,27:807-810
[3]Nasr M N A,Said S A.Arch.Pharm.Med.Chem.,2003,336:551-559
[4]Gupta S,Barik A K,Pal S,et al.Polyhedron,2007,26:133-141
[5]Özdemir Z,Kandilci H B,Güml B,et al.Eur.J.Med.Chem.,2007,42:373-379
[6]WEI Dong-Ming(魏 东 明),DONG Bin(董 斌),LI Zhang-Peng(李章鹏),et al.Chinese J.Inorg.Chem.(Wuji Huaxue Xuebao),2011,27(5):891-897
[7]Fujisawa K,Lehnert N,Ishikawa Y,et al.Angew.Chem.Int.Ed.,2004,43:4944-4947
[8]Dias H V R,Singh S,Campana CF.Inorg.Chem.,2008,47:3943-3945
[9]WANG Xin-Yu(王欣羽),LI Zhen(李桢),SUN Qiao(孙巧),et al.Chinese J.Inorg.Chem.(Wuji Huaxue Xuebao),2012,28(3):483-490
[10]XING Yong-Heng(邢永恒),ZHANG Yuan-Hong(张元红),XU Yong-Ting(徐永廷),et al.Chin.Sci.Bull.(Kexue Tongbao),2006,51:2189-2196
[11]Day J,Marriott K E R,Kilner C A,et al.New J.Chem.,2010,34:52-60
[12]Adams CJ,Haddow MF,Hughes RJI,et al.Dalton Trans.,2010,39:3714-3724
[13]Casarin M,Corvaja C,Nicola C D,et al.Inorg.Chem.,2004,43:5865-5876
[14]Nicola C D,Garau F,Gazzano M,et al.Cryst.Growth Des.,2010,10:3120-3131
[15]Dias H V R,Polach SA,Wang Z.J.Fluorine Chem.,2000,103:163-169
[16]Carrillo M R,Chakraborty I,Raptis R G.Cryst.Growth Des.,2010,10:2606-2612
[17]Sheldrick G M.SHELX-97,Program for Crystal Structure Analysis,University of Gottingen,Gottingen,Germany,1997.
[18]Fujisawa K,Iwamoto H,Tobita K,et al.Inorg.Chim.Acta,2009,362:4500-4509
[19]Dias H V R,Diyabalanage H V K,Eldabaja M G,et al.J.Am.Chem.Soc.,2005,127:7489-7501
[20]Hu M H,Shen G L,Zhang J X,et al.Cryst.Growth Des.,2009,9:4533-4537
[21]Frisch M J T,Schlegel G W,Scuseria H B,et al.Gaussian 09,Revision A.1,Gaussian,Inc.,Wallingford CT,2009.
[22]Dreizler R M,Gross E U K.Density Functional Theory.Heidelberg,Germany:Springer-Verlag,1990.
[23]Lee C T,Yang W T,Parr R G.Phys.Rev.B,1988,37:785-789
[24]Delley B.J.Chem.Phys.,1990,92:508-517
[25]Delley B.J.Chem.Phys.,2000,113:7756-7764
[26]Perdew JP,Wang Y.Phys.Rev.B,1992,45:13244-13249
猜你喜欢
高中数理化(2022年16期)2022-09-14
今日农业(2021年2期)2021-11-27
今日农业(2020年23期)2020-12-31
青岛大学学报(工程技术版)(2019年2期)2019-09-10
中学生数理化(高中版.高考理化)(2019年6期)2019-06-22
枣庄学院学报(2015年5期)2016-01-09
中学化学(2015年8期)2015-12-29
无机化学学报(2014年9期)2014-02-28
无机化学学报(2014年5期)2014-02-28
火炸药学报(2012年4期)2012-01-29
