
微波固相法快速制备氮掺杂石墨烯
2013-09-15 03:03季炳成韩美佳李立伟
无机化学学报 2013年10期
王 凯 季炳成 韩美佳 李立伟
(1青岛大学自动化工程学院,青岛 266071)
(2大连理工大学电气工程学院,大连 116023)
(3中国市政工程西南设计研究总院)
石墨烯是由sp2碳原子构筑而成的具有单原子层厚度的二维晶体材料,是继富勒烯和碳纳米管发现之后的又一种新型的碳同素异构体[1-3]。这种独特的结构使其表现出传统材料所不具有的多种非凡性能[4-5],比如优异的化学稳定性和较大的比表面积,出众的导热性和导电性[6],使其成为极具前景的超级电容器电极材料[7-10]。若对石墨烯进行氮掺杂,能够有效地改变石墨烯的电子传输特性以及表面润湿性,从而进一步提升其用作电极材料时的性能[11]。
制备氮掺杂石墨烯主要通过直接合成以及后处理等两条途径。直接合成是指在化学气相沉积合成石墨烯时通入氮源使氮原子在石墨烯生长的过程中原位掺杂进入其晶格;而后处理则是在高温下利用含氮分子对石墨烯进行处理,使氮原子掺杂进入到石墨烯的晶格中[12-15]。目前,研究者已实现利用氧化石墨为原料大规模制备石墨烯,因此后处理掺氮的途径更具有实际应用的潜力[16]。为了实现对石墨烯的高效氮掺杂,具有较高反应性的NH3常被选作氮源[17]。但氨气有毒并且具有强烈的腐蚀性,对设备提出了很高的要求,且不利于实现大规模的制备。鉴于此,本研究提出一种利用乙二胺做为氮源的固相微波辅助快速高效制备氮掺杂石墨烯的方法,即利用乙二胺与氧化石墨上环氧键间的开环反应首先得到功能化石墨烯,随后将干燥后的功能化石墨烯进行微波处理,实现对石墨烯的深度还原以及氮掺杂。
1 实验部分
1.1 氧化石墨的制备
在1 000 mL的烧杯中加入130 mL浓硫酸和5 g鳞片石墨,并在冰浴条件下机械搅拌2 h。向上述体系中缓慢添加15 g高锰酸钾,并维持在冰浴条件下继续搅拌2 h,这一阶段为低温反应阶段。撤去冰浴,在35℃水浴下继续搅拌1 h,进一步促进石墨的氧化,该阶段为中温反应阶段。缓慢地向烧杯中滴加230 mL去离子水,并升温至98oC继续反应30 min,此阶段为高温反应阶段。高温反应结束后其中加入400 mL去离子水,并离心洗涤至中性得到氧化石墨分散液。
1.2 氮掺杂石墨烯的制备
将氧化石墨分散液稀释至1 mg·mL-1的浓度,量取120 mL该分散液并与1.5 mL乙二胺加入至250 mL单口烧瓶于95℃下回流反应6 h,反应完成后通过冷冻干燥得到固相FGS(功能化石墨烯)。将所得到的FGS置于微波炉中,在Ar气氛下满功率微波处理1 min后得到氮掺杂石墨烯。
2 结果与讨论
2.1 氮掺杂石墨烯的SEM和EDS分析
图1(a)(b)(c)所示分别为氧化石墨、功能化石墨烯、氮掺杂石墨SEM图像。从图1(a)和(b)中可以看出,氧化石墨呈紧密的堆积状态,这主要是由于较强的层间范德华力所致。经过功能化和微波处理之后,片层之间的相互堆叠程度明显减弱,表明片层发生了明显的剥离。此外,图1(c)中剥离的片层呈无序堆叠,表面呈现出丰富的褶皱。说明通过功能化以及微波处理的途径能充分将氧化石墨剥离成片层很薄的石墨烯结构。
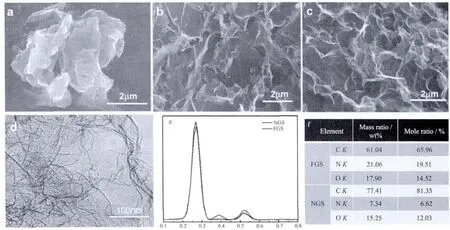
图1 GO,FGS,NGS扫描电镜与透射电镜图片和EDS分析Fig.1 SEM and TEM images of GO,FGSand NGSas well as EDSanalysis of FGSand NGS
图1 (d)所示为氮掺杂石墨烯的TEM图像。如图1(d)所示,氮掺杂石墨烯呈无序、透明、褶皱的薄纱状,部分薄片层叠在一起,形成多层结构。这种边缘卷曲褶皱的形貌,可能来源于氮原子掺杂进石墨烯晶格所造成的缺陷结构[18]。
图1(e)为功能化石墨和氮掺杂石墨烯的EDS分析,其中各种元素的质量比以及物质的量比列于图1(f)中。由图1(d)可见,功能化石墨和氮掺杂石墨烯分别在 0.27、0.39、0.52 keV 处出现 C1s、N1s、O1s峰。对比两条曲线可见,氮掺杂石墨烯C1s峰强度明显升高,O1s和N1s峰强度则明显降低,这是因为微波辐照使温度迅速升高,功能化石墨中的含氧极性官能团迅速分解成CO、CO2和H2O,而接枝上的乙二胺分子也会在高热作用下分解脱离石墨烯表面,同时在极高的温度下这些含氮的分子与石墨烯发生反应,实现对石墨烯的掺杂。由图1(e)可知,C/O原子比由反应前的3.41%(质量比)变为反应后的5.08%(质量比),并且氮元素含量为7.34%(质量比),说明实验成功地进行了氮掺杂。
2.2 氮掺杂石墨的红外图谱
图2为氧化石墨、功能化石墨烯和氮掺杂石墨烯的FTIR图谱。从图中可以看出,GO在1 732 cm-1和1 050 cm-1处存在较强的吸收峰,分别对应C=O键和C-O的伸缩振动。1 614 cm-1处的吸收峰为芳环骨架的振动。与乙二胺反应后,C-O的伸缩振动明显减弱,同时在1 471 cm-1和1 534 cm-1处出现了明显的N-H吸收峰,说明乙二胺分子成功接枝到石墨烯表面。经过微波处理,有关含氧官能团的特征吸收大幅减弱,说明微波处理过程能有效的脱除GO表面的含氧官能团。
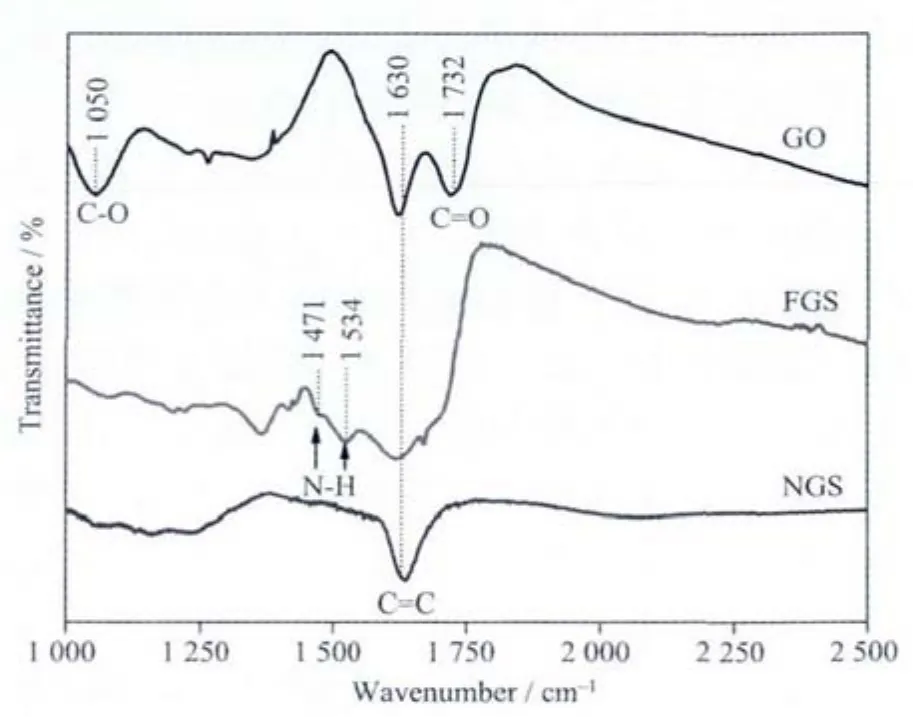
图2 GO、FGS和NGS的FTIR图谱Fig.2 FTIR spectra of GO,FGSand NGS
2.3 氮掺杂石墨烯的XPS光谱
为了进一步分析产物中N元素的存在状态,对其进行了XPS分析。FGS以及NGS的全谱中都表现出了位于284.6 eV的C1s峰,399 eV的N1s峰以及531.8 eV的O1s峰,并且NGS中N和O对应峰的强度明显弱于FGS中相应峰的强度,这与EDS表征的结果是一致的。由于碳材料的化学稳定性很高,因此要实现对其进行掺杂处理往往需要较高的温度。因此,通过乙二胺对氧化石墨进行处理后,其N1s高分辨谱图没有表现出掺杂形态的氮元素,而主要是以含胺基官能团的形式存在,这与文献上报道的结果是一致的[19-20]。经微波处理之后,微波与石墨烯之间的强烈的相互作用会在瞬间产生极高的温度,使FGS表面的大部分官能团发生脱落,同时胺基热降解产生的部分分子或自由基在这个过程中可能掺杂进入到了石墨烯的晶格中[21]。从NGS的高分辨N1s谱图中可以看出,产物中的氮元素主要以3种形式存在,分别为位于398.2 eV的吡啶型氮,399.7 eV的吡咯型氮以及401.3 eV的石墨型氮,表明氮元素成功掺杂进入到石墨烯的晶格中。
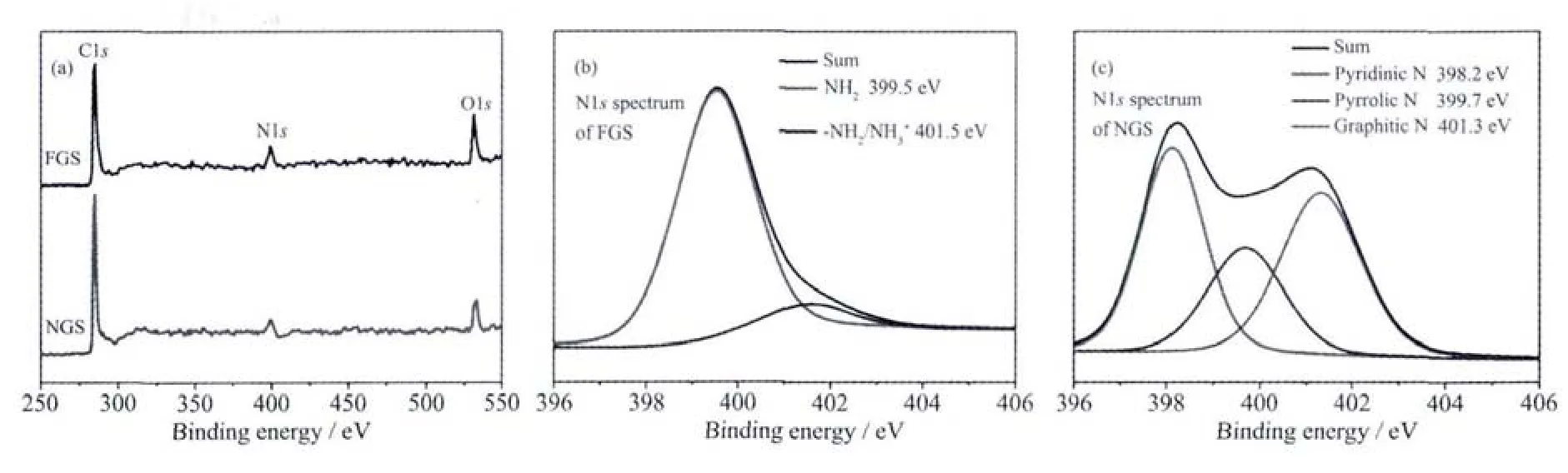
图3 FGS和NGS的XPS全谱,以及FGS和NGS的N1s谱Fig.3 XPSspectra of FGSand NGS,N1s spectra of FGSand NGS
2.4 氮掺杂石墨烯X射线衍射(XRD)
图3 是氧化石墨、功能化石墨烯和氮掺杂石墨烯的XRD图。从图中可以看出,GO(001)衍射峰出现在2θ=11.48°处,说明成功的对天然鳞片进行了插层,这将有助于减小片层之间的相互作用力,有助于进一步的对片层剥离。经乙二胺还原处理后,生成的FGS中GO(001)衍射峰消失,说明乙二胺成功的接枝到氧化石墨烯的表面,削弱了层间的范德华力,有效阻止了片层之间的重新堆叠。此外,FGS在20°~30°之间变现出很宽的衍射峰,说明片层之间堆积基本呈现无序状态。经过微波加热后,NGS在20°~30°之间的衍射峰相对于FGS有所增强,并且峰中心位置向右偏移,但仍偏离鳞片石墨(002)衍射峰位置,这可能是由于剥离以及掺杂等过程在石墨烯表面造成的起伏和褶皱对其重新堆叠的阻碍作用。
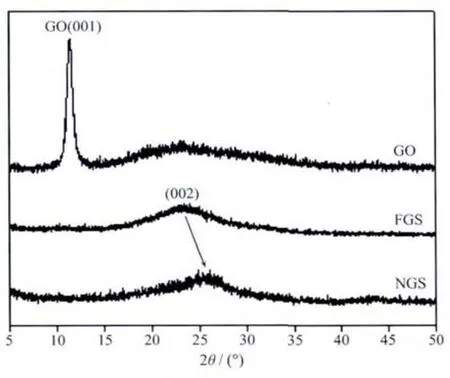
图 3 GO、FGS和 NGS的 XRD图Fig.3 XRDpatterns of GO,FGSand NGS
2.5 氮掺杂石墨烯的Raman表征
图 4为 GO、FGS和 NGS的 Raman光谱对比图。两条谱线在1 344和1 590 cm-1附近都有2个明显的谱峰,分别对应石墨烯的D模和G模。在经历了剧烈的氧化插层过程后,GO相对于天然鳞片石墨表现出了很高的ID/IG值[22-25]。对GO的乙二胺功能化处理之后引入了更多的sp3碳原子,使得FGS的发生了进一步的提高,由0.97增加到了1.25。经过微波辐照处理,NGS的D模强度有所下降,其ID/IG值为1.16,这主要是因为在微波作用下产生的高热过程能有效的脱除表面的功能化基团,起到了有效的还原作用。
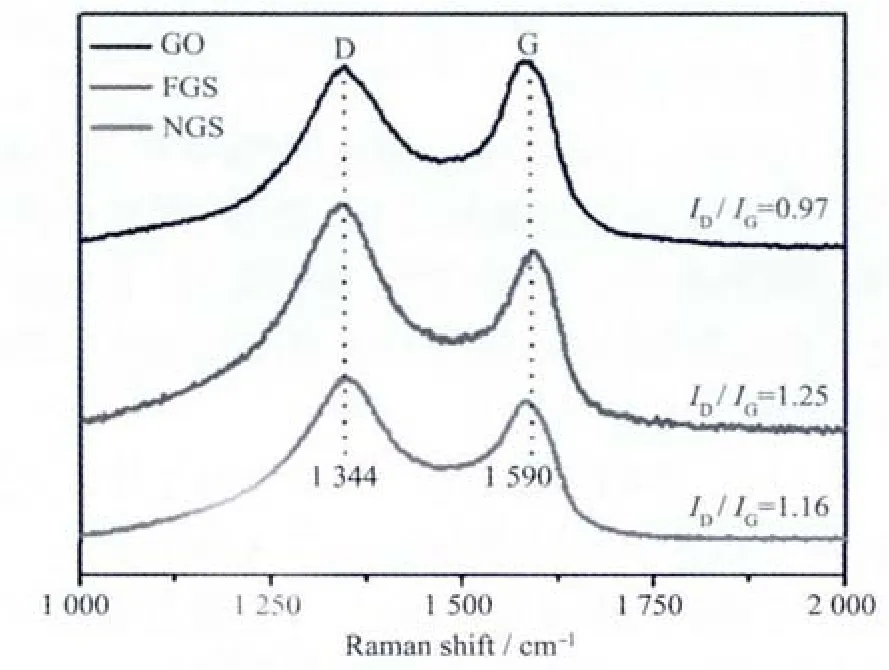
图4 GO、FGS和NGS的Raman光谱Fig.4 Raman spectra of GO,FGSand GS
2.6 微波固相法合成氮掺杂石墨烯
氧化石墨表面存在大量的环氧官能团,能够与乙二胺的胺基发生开环反应,使乙二胺分子接枝到片层表面。干燥后的功能化石墨烯由于存在大量未被完全氧化共轭区域,该区域能强烈吸收微波,在微波的作用下瞬间产生高温,从而导致含氧官能团脱离石墨烯表面。与此同时,接枝到表面的乙二胺分子也会在高温的作用发生分解,并掺杂到石墨烯的晶格中,成功实现对石墨烯的掺杂。
3 结 论
采用乙二胺对氧化石墨进行功能化,并通过固相微波处理乙二胺功能化石墨烯的过程成功实现对石墨烯的氮掺杂。相对于传统的氮掺杂方法,该方法避免了使用高腐蚀性以及高毒性的氨气,表现出环境友好的特点。此外,这种方法还兼具高效以及高掺杂量等特点。
[1]Guo SJ,Dong SJ.Chem.Soc.Rev.,2011,40:2644-2672
[2]Meyer J C,Geim A K,Katsnelson M I,et al.Nature,2007,446:60-63
[3]Wang K,Zhang L.Int.J.Electrochem.Sci.,2013,8:2892-2897
[4]Reddy A,Srivastava A,Gowda SR,et al.ACSNano,2010,4:6337-6342
[5]Wang K,Zhang L.Electrochemistry,2013,81:259-261
[6]Ohta T,Bostwick A,Seyller T,et al.Science,2006,313:951-954
[7]Novoselov K S,Geim A K,Morozov S V,et al.Science,2004,306:666-669
[8]Wang Y,Shao Y Y,Matson D W,et al.ACSNano,2010,4:1790-1798
[9]Xuan W,Lin JZ,Mullen K.Nano Lett.,2008,8:323-327327
[10]Stoller M D,Park SJ,Zhu Y W,et al.Nano Lett.,2008,8:3498-3502
[11]Wang X R,Li X L,Zhang L,et al.Science,2009,324:768-771
[12]YANG Yong-Hui(杨勇辉),SUN Hong-Juan(孙红娟),PENG Tong-Jiang(彭同江).Chinese J.Inorg.Chem.(Wuji Huaxue Xuebao),2010(11):2083-2090
[13]SU Peng(苏鹏),GUO Hui-Lin(郭 慧林),PENG San(彭三),et al.Acta Phys.-Chim.Sin.(Wuli Huaxue Xuebao),2012(11):2745-2753
[14]Panchokarla L S,Subrahmanyam K S,Saha SK,et al.Adv.Mater.,2009,21:4726
[15]Liu R L,Wu D Q,Feng X L,et al.Angew.Chem.Int.Ed.,2010,49:2565-2569
[16]Shen WZ,Fan W B.J.Mater.Chem.A,2013,1:999-1013
[17]Li X L,Wang H L,Robinson JT,et al.J.Am.Chem.Soc.,2009,131:15939-15944
[18]Sheng Z H,Shao L,Chen J J,et al.ACS Nano,2011,5:4350-4358
[19]Compton O C,Dikin D A,Putz K W,et al.Adv.Mater.,2010,22:892-896
[20]Che J,Shen L,Xiao Y.J.Mater.Chem.,2010,20:1722-1727
[21]Hu H,Zhao Z,Zhou Q,et al.Carbon,2012,50:3267-3273
[22]MAGui-Xiang(马贵香),ZHAOJiang-Hong(赵江红),ZHENG Jiang-Feng(郑剑锋),et al.New Carbon Mater.(Xinxing Tan Cailiao),2012,27(4):258-265
[23]Hu H,Zhao Z,Wan W,et al.Adv.Mater.,2013,25:2219-2223
[24]Chen W F,Yan L F,Bangal P R.Carbon,2010,48:1146-1152
[25]Zhu Y W,Murali S,Stoller M D,et al.Carbon,2010,48:2118-2122
猜你喜欢
中学生数理化·高一版(2022年4期)2022-05-09
节能与环保(2022年3期)2022-04-26
含能材料(2022年4期)2022-04-16
中学生数理化(高中版.高考数学)(2020年2期)2020-04-21
中国材料进展(2019年10期)2019-12-07
通信电源技术(2016年6期)2016-04-20
当代化工研究(2016年5期)2016-03-20
高中生学习·高二版(2015年3期)2015-05-21
应用化工(2014年1期)2014-08-16
江苏农业科学(2014年4期)2014-07-11
