
重叠延伸PCR技术构建β-环糊精葡糖基转移酶基因的定点突变原核表达载体
2013-09-04 10:21于寒松朴春红王玉华刘俊梅胡耀辉
食品工业科技 2013年19期
王 华,文 一,于寒松,朴春红,王玉华,刘俊梅,胡耀辉,*
(1.内蒙古民族大学生命科学学院,内蒙古通辽028000;2.吉林农业大学食品学院,吉林长春130118;3.环境保护部环境规划院,北京100012)
β-环糊精葡萄糖基转移酶(β-CGTase)属于α-淀粉酶家族,可以作用于淀粉的葡萄糖单位通过α-1,4-糖苷键连接而生成 β-环糊精(β-CD)。β-CD的化学结构具有非极性空穴和外亲水的特点,常被用作分子胶囊和乳化剂,广泛应用于食品、医药、化妆品、农药等领域[1-2]。
β-CGTase是催化淀粉发生环化反应生成β-CD的关键酶,但是酶法生产由于酶活性的影响使得产率低[3]。近年来,基因工程定点突变技术广泛应用于酶的体外改造中,通过改变酶的基因序列来改变酶与底物的结合能力,从而改变酶的活性。Shin Hyun-Dong等[4]人利用定点突变技术将 β-CGTase基因中W652突变成G,结果表明酶的环化活性增强而水解及偶合活性降低。通过对来自B,circulans 251中的环糊精转移酶进行突变Ala230Val,使该酶的水解活性提高而环化活性降低[5]。重叠延伸 PCR技术(overlap extension PCR)是1989年Horton等人建立的一种PCR扩增方法[6],它通过寡聚核苷酸链之间相互重叠的部分搭桥,互为模板,通过多次PCR扩增,从而获得目的DNA基因片段的方法。重叠延伸PCR技术可以准确、高效地扩增DNA片段,尤其在进行定点突变时可以提高突变效率,并且不受突变位置及突变类型的限制[7]。本实验通过在SWISSMODEL上建模,根据蛋白质三维结构,考虑到β-CGTase属于α-淀粉酶家族,其基因序列C端的保守区是与淀粉酶的特异性和稳定性有关,不影响酶活性质,因此将N端典型保守区域氨基酸残基进行改变,从而改变突变位点的疏水性及其空间结构[8],分析得到β-CGTase保守区内可能影响到酶环化活性的二个关键位点 Y127和 R254,采用重叠延伸PCR技术进行定点突变,为β-CGTase环化活性的提高提供研究基础。
1 材料与方法
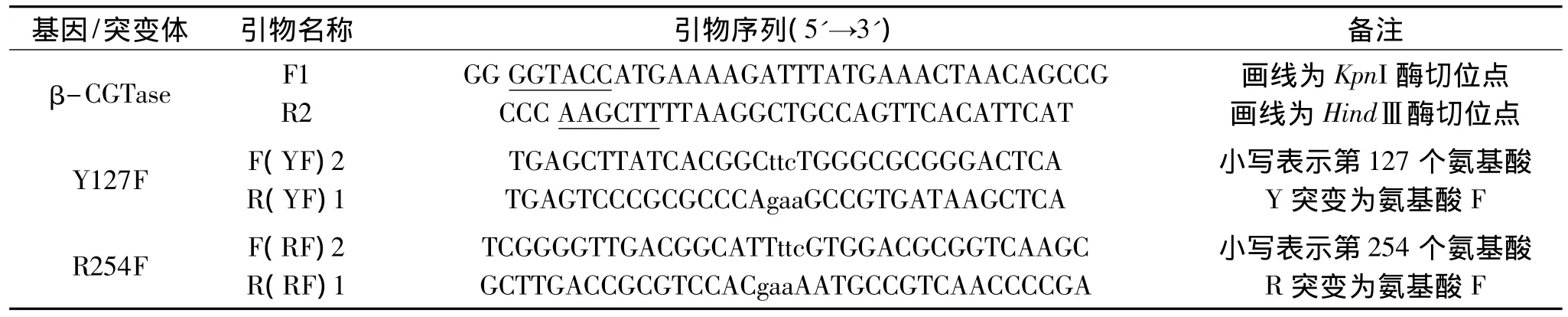
表1 重叠延伸PCR的引物Table 1 Primer pairs of overlap extension PCR

表2 引物配对和PCR产物Table 2 Primer pairs and production of PCR
1.1 材料与仪器
菌株Escherichia coli BL21(DE3)上海光明乳业有限公司技术中心惠赠;重组表达质粒 pET-28b::CGTase 上述技术中心构建;Primer STAR HS和Taq聚合酶、克隆载体pMD18-T Simple、表达载体pUC-18,T4 DNA连接酶、限制性内切酶 HindⅢ和KpnⅠ 均购自TaKaRa公司;氨苄青霉素贮存液 氨苄青霉素 北京鼎国,用无菌超纯水配成100mg/mL,分装,-20℃冻存,使用时每1mL培养基加入lμL,终浓度为100μg/mL;常规化学试剂 均为国产分析纯试剂;液体 LB 培养基[9]1.0% 胰蛋白胨,1.0%NaCl,0.5%酵母浸出物,加蒸馏水充分溶解后,定容至1L,分装,高压蒸汽灭菌118℃,20min。
PCR扩增仪 ABI,Applied Biosystems;凝胶成像系统 DNr MiniBis pro。
1.2 实验方法
1.2.1 重叠延伸PCR 扩增原理[10]如图1所示,F1和R2与原模板完全匹配,F2和R1是引入的突变位点,并且具有12个以上相匹配的互补碱基。以ORF为模板,用引物F1和R1,F2和R2分别扩增得到ORF-1和 ORF-2,再以 ORF-1和 ORF-2 混合物为模板,用引物F1和R2扩增得到突变的基因序列。
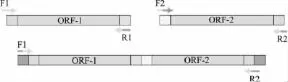
图1 重叠延伸PCR原理Fig.1 Principle of overlap extension PCR
1.2.2 引物 根据 β-CGTase基因和2个突变氨基酸相应位点及其两侧的碱基序列,用软件 Primer premier 5.0设计一对β-CGTase的引物和2对突变引物,如表1。
1.2.3 PCR 扩增 第一轮 PCR:以质粒 pET-28b::CGTase为模板,根据重叠延伸PCR的反应原理,设定扩增ORF-1和ORF-2片段程序:94℃ 5min;94℃45s,55℃ 45s,72℃ 90s,35 个循环;72℃ 10min;4℃保温。
第二轮PCR:以 ORF-1和 ORF-2混合物为模板,扩增得到突变的基因片段程序:94℃ 5min;94℃45s,55℃ 45s,72℃ 2min,35 个循环;体系中加入 Taq聚合酶,72℃ 30min;4℃保温[11]。
1.2.4 突变目的基因的鉴定 PCR扩增后,取5μL PCR产物用于1.0%的琼脂糖凝胶电泳检测。
1.2.5 克隆载体的转化与鉴定 鉴定正确的突变基因使用TIGEN胶回收试剂盒进行PCR产物纯化回收,经过纯化的产物与pMD18-T Simple载体连接。利用热击法将克隆载体转化到E.coli DH5α中,在含有终浓度为100μg/mL氨苄青霉素的LB平板上进行蓝白斑筛选,挑取白色单菌落进行液体扩大培养,经过菌液PCR检测及质粒双酶切鉴定片段大小正确后,将阳性菌株送上海生工进行基因测序。
1.2.6 原核表达载体的构建 将已确认的pMD18-T重组质粒和pUC-18表达载体用限制性内切酶KpnⅠ和HindⅢ分别进行双酶切,纯化回收目的基因和表达载体并使用T4 DNA连接酶将其连接,连接产物转化E.coli BL 21(DE3),在含有终浓度为100μg/mL氨苄青霉素的LB平板上进行蓝白斑筛选,得到阳性菌株,进行酶切鉴定和DNA序列测定。
2 结果与讨论
2.1 PCR扩增结果
2.1.1 第一轮PCR:ORF-1和ORF-2片段扩增 用适当稀释的质粒pET-28b::CGTase为模板,以表2所示的引物进行配对PCR扩增,分别获得相应的PCR产物。经1.0%的琼脂糖凝胶电泳检测,结果与预计相符,并且没有非特异性条带,结果如图2所示。
2.1.2 第二轮PCR突变基因片段扩增 第一次获得的各突变体的ORF-1和ORF-2基因片段纯化后经适当稀释等量混合作为模板,用引物F1/R2进行扩增,经1.0%琼脂糖电泳检测,产物大小均为2139bp,且无非特异性条带,结果与预计相符,如图3。
2.2 PCR产物鉴定
与T-vector连接后转化到 E.coil DH5α 中,经过菌液PCR鉴定,结果正确后再提取质粒,用HindIII和Kpn I进行双酶切(图4),酶切片段大小正确,再进行测序鉴定。
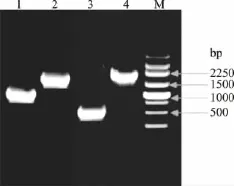
图2 不同突变体的ORF-1和ORF-2片段扩增电泳分析Fig.2 Electrophoresis of ORF-1 and ORF-2 amplified fragment of different mutants
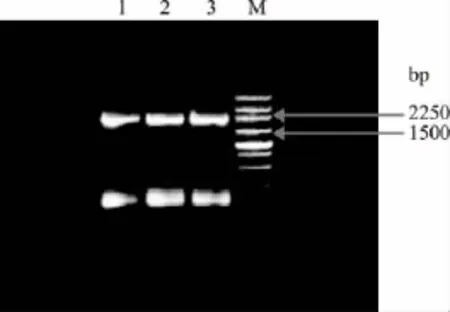
图3 不同突变体的突变基因片段扩增电泳分析Fig.3 Electrophoresis of the amplified fragment of different mutants
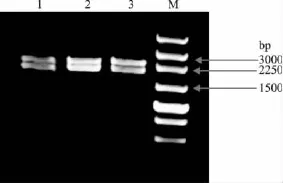
图4 突变体双酶切鉴定Fig.4 Restriction enzyme digestion of the mutants
2.3 克隆载体的测序分析
利用clustalx1.81对原基因β-CGTase蛋白序列和二个突变体Y127F,R254F的蛋白序列进行多序列比对,所获得突变基因是预先设计的定点突变(图5),表明β-CGTase突变体构建成功同时也说明了overlapPCR技术是一种简便且有效的定点突变方法。
2.4 原核表达载体的构建

图5 β-CGTase基因序列与突变基因序列多重序列比较Fig.5 Multiple alignment of the amino-acid sequences of β-CGTase and mutants
经上海生工测序,测序结果与克隆载体测序一致,表明突变的目的基因已插入到pUC18表达载体中,成功构建了原核表达载体。
3 结论
3.1 本研究采用重叠延伸PCR技术成功地将β-CGTase基因活性中心关键位点的两个氨基酸Y127,R254分别进行定点突变,得到突变体Y127F和R254F,构建出突变重组表达载体,并转化至大肠杆菌中诱导表达,使β-CGTase基因实现了在大肠杆菌中的有活力表达,为后续β-环糊精葡糖糖基转移酶的环化活性提高提供研究基础。
3.2 重叠延伸PCR技术可以将两个DNA片段连接在一下,使其能够在体外进行准确、高效的基因重组,且不需要使用内切酶消化和连接酶处理。重叠延伸PCR技术成功的关键是重叠互补引物的设计以及模板的纯度,重叠互补引物3'端与模板作用,5'端与重叠延伸中待融合的DNA片段互补,引物重叠部分应有15~20个碱基,突变的碱基设计在引物的中间部位。除此之外,还要考虑引物Tm值对PCR结果的影响,引物的GC含量对Tm值的影响很大,因此引物设计时尽量降低引物中GC的含量;一个PCR体系中的所有引物的Tm值应该相近,以确保延伸扩增的顺利进行。模板的纯度主要是指第二轮PCR中使用第一轮PCR产物作为模板,因此产物需经过纯化,通常当模板 OD260/OD280 为 1.75~1.80 为最佳。本实验应用Primer premier 5.0软件设计突变引物,突变碱基设计在引物的中间部位,引物重叠部分在15~20个碱基之间,引物GC含量在40%~60%之间,第一轮PCR产物经切胶回收浓度达到50ng/μL做为第二轮PCR反应的模板,经延伸PCR扩增产物测序证实在位点Y127,R254成功地进行了基因突变。
[1]Rao P,Suresh C,Rao D Narasimha,et al.Digestion of residual β-cyclodextrin in treated egg using glucoamylase from a mutant strain of Aspergillusniger[J].Food Chemistry,1999,65:297-301.
[2]田辉,杨国武,徐颐玲,等.环状糊精与环状糊精葡萄糖基转移酶[J].工业微生物,1995,25(2):33-38.
[3]王媛,王荫榆,郭本恒,等.表达环糊精葡基转移酶基因的重组大肠杆菌发酵条件的研究[J].工业微生物,2007,37(3):10-14.
[4]Shin H D,Park T H,Lee Y H,et al.Site- directed mutagenesis and functional analysis of maltose-binding site of β-cyclodextrin glucanotransferase from Bacillusfirmusvar.alkalophilus[J].Biotechnology Letters,2000(22):115-121.
[6]Leemhuis H.Engineering cyclodextrin glycolsyltransferase into a starch hydrolase with a high exo- specificity[J].J Biotechnol,2003,103(3):203-212.
[7]Horton R M,Pullen J K,Pease L R,et al.Site- directed mutagenesis by overlap extecsion using the Polymerase chain reaction [J].Gene,1989,77(1):51-59.
[8]姚婷,李华钟,房耀维,等.定点突变提高 Thermococcus siculi HJ21高温酸性α-淀粉酶的催化活性[J].食品科学,2011,32(15):148-152.
[9]Mergulhão F J,Kelly A G,Monteiro G A,et al.Troubleshooting in gene splicing by overlap extension:a step-wise method[J].Mol Biotechnol,1999,12(3):285-287.
[10]黄培堂,俞炜源,陈添弥,等.PCR技术实验指南[M].北京:科学出版社,1999.416-444.
[11]Urban A,Neukirchen S,Jaeger KE,et al.A rapid and efficient method for site-directed mutagenesis using one-step overlap extension PCR [J].Nucleic Acids Res,1997,25(11):2227-2228.
猜你喜欢
中学生数理化(高中版.高考数学)(2022年4期)2022-05-25
今日农业(2021年21期)2021-11-26
新世纪智能(教师)(2021年2期)2021-11-05
教育周报·教育论坛(2021年21期)2021-04-14
中成药(2018年8期)2018-08-29
中成药(2018年6期)2018-07-11
生命科学研究(2018年1期)2018-05-29
中成药(2018年4期)2018-04-26
安徽医科大学学报(2016年12期)2017-01-15
天津医科大学学报(2015年2期)2015-12-22
