
基于细菌酯酶活性的叠氮噻唑橙分子设计与特性验证*
2013-08-16 05:47:38余以刚黄韵李晓凤肖性龙吴晖
华南理工大学学报(自然科学版) 2013年10期
余以刚 黄韵 李晓凤 肖性龙 吴晖
(华南理工大学轻工与食品学院,广东广州510640)
食源性致病菌检验的关键是食品中“活”的致病菌的存在性检测和准确定量[1].目前用于区别活菌和死菌的标准主要有:是否可培养、是否有代谢活性以及细胞膜是否完整.分离培养法对于活的非可培养状态的细菌会出现漏检[2].EMA/PMA-qPCR活菌检测技术利用新型核酸染料EMA(叠氮溴化乙锭)或PMA(叠氮溴化丙锭)渗入膜损伤的细胞后,与DNA发生共价结合,从而抑制膜损伤细胞DNA在PCR反应中的扩增[3].该方法实际上是通过细胞膜的完整性来对“死”、“活”细胞进行区分检测[4].有研究表明,这种活菌PCR技术在方法和原理上仍存在不足[5-8].细胞膜损伤可能是细菌死亡的一种极端表现形式,以细胞膜的完整性作为区分死、活菌的检测标准存在缺陷.因此,建立一套基于生物代谢活性的活菌分析方法,使其既可有效检测出活菌(包括VBNC菌),又可克服EMA/PMA-qPCR方法在原理上的不足,对于膜完整或膜损伤的死细胞不检测,这也符合致病菌检测的根本要求.
为了通过细胞内有无代谢活性来区分活、死菌,达到更精确的致病菌活菌检测的目的,笔者设计了一种对细菌生物代谢活性敏感的新型噻唑橙荧光染料(简称TOMA),它主要由3个功能基团构成:一是噻唑橙基团(使分子可以自由穿透细胞),二是叠氮基团(在光照下与核酸共价结合后抑制核酸扩增),三是连接两个基团并含有一个酯键的柔性碳链(酯键对生物酯酶活性敏感,可被酯酶水解断裂).TOMA选取了具有细胞穿透性的菁染料作为DNA结合基团,故可进入所有细胞.利用带酯键的碳链,可将染料基团与叠氮基团连接起来,而在酯酶水解作用下,两个基团可相互分离.在活性细胞内,TOMA分子中的碳链被酶解断裂,叠氮基团从分子上脱落;反之,在无活性的细胞内,叠氮基团不会脱落,在可见光的作用下与 DNA共价交联形成共价化合物,抑制DNA后续扩增.而游离的TOMA分子,其叠氮基团因在可见光作用下与水反应生成羟胺而被钝化.因此,结合TOMA与定量PCR(qPCR)技术建立新型活菌检测技术来区分活、死菌,将依赖于细胞体内的酯酶活性,而不是细胞膜完整性,理论上可比以往的活菌检测技术更准确.为建立以“细菌酯酶代谢活性”作为判断细菌存活标准的活菌检测技术,文中通过人工设计合成了TOMA分子,并尝试在实验室条件下对TOMA分子的预期功能特性进行了体外验证.
1 材料与方法
1.1 实验材料
1.1.1 实验菌株与培养条件
实验所用菌株为金黄色葡萄球菌(Staphyloccocus aureus)(ATCC6538)、大肠杆菌(Escherichia coli)O157:H7(ATCC6589)以及单增李斯特菌(Listeria monocytogenes)(CMCC34761),均为实验室保存菌种.
细菌培养采用LB液体培养基(蛋白胨10 g/L,酵母提取物 5g/L,NaCl 10 g/L),pH 值调至 7.0,培养基经过121℃、20min灭菌后使用.将细菌分别接种到30 mL LB液体培养基中,于37℃下180 r/min摇床中过夜培养16 h(OD600≈1).液体培养基中添加1.5%(质量分数)的琼脂粉即得相应的固体培养基,用于平板法测可培养菌数.
1.1.2 试剂及仪器
所用试剂均为分析纯或色谱纯;固定化脂肪酶Novozym435(固定化于大孔丙烯酸树脂,酶活力10U/mg)购自丹麦 NovoNordisk Industries公司;细菌基因组DNA快速提取试剂盒购自北京艾德莱生物科技有限公司;PCR AmpLification Kit购自TaKaRa公司.
Waters 1525-ZQ2000型液相质谱联用仪、高效液相色谱仪、2996 PDA型检测器,美国沃特世公司生产;Bruker Biospin AG AV 600型核磁共振仪,瑞士布鲁克拜厄斯宾有限公司生产;Laica SP5型激光共聚焦显微镜,德国徕卡微系统有限公司生产;卤钨灯,650W,购自德国Osram公司;台式高速冷冻离心机,德国艾本德股份有限公司生产;水浴锅购自新加坡雷蒙特实验设备公司;ABI7500型荧光定量PCR仪,购自美国应用生物系统公司.
1.2 TOMA染料的合成
TOMA分子的合成路线如图1所示,委托上海辉睿生物技术有限公司合成.目标物经合成与纯化后,使用高分辨质谱仪与核磁共振仪对分子结构进行确证,粉末于4℃下避光保存.1H NMR(600MHz,d6-DMSO):δ1.33 ~1.42(m,2H,CH2),1.54 ~ 1.63(m,2H,CH2),1.84 ~ 1.89(m,2H,CH2),2.28 ~2.36(t,2H,CH2),3.38 ~3.43(t,2H,CH2),4.14 ~4.18(t,2H,CH2),4.57 ~4.63(t,2H,CH2),6.92 ~6.96(s,1H,═CH),7.34~7.38(d,1H,Ar-H),7.40~7.44(t,1H,Ar-H),7.57 ~7.63(t,1H,Ar-H),7.72 ~7.76(t,1H,Ar-H),7.77 ~7.80(d,1H,Ar-H),7.95 ~8.01(t,1H,Ar-H),8.04 ~8.08(d,1H,Ar-H),8.11 ~8.14(d,1H,Ar-H),8.62 ~8.66(d,1H,Ar-H),7.87 ~8.83(d,1H,Ar-H).

图1 目标化合物TOMA的合成路线Fig.1 Synthetic route of target compound TOMA
1.3 TOMA分子功能验证
1.3.1 TOMA对细菌胞膜的穿透性验证
称取50mg TOMA粉末溶解于1 mL 20%(体积分数)的二甲基亚砜(DMSO)中,得到50 mg/mL的储备液.向90μL无菌超纯水中加入10μL TOMA储备液,混匀,得到5mg/mL的TOMA工作液.
分别取500μL新鲜培养的金黄色葡萄球菌、大肠杆菌O157:H7以及单增李斯特菌菌液于1.5 mL离心管中,6000 r/min离心3 min,等体积无菌水重悬.菌液中加入TOMA工作液1μL使菌液中TOMA终浓度为10 μg/mL,充分混匀后,于室温下暗处孵育10min.将离心管水平放置于冰上,于650W的卤钨灯下方20cm处进行5min光照,期间轻轻摇晃冰盒保证溶液得到充分照射.阴性对照组使用无菌超纯水代替TOMA溶液.
对经TOMA处理后的菌体使用10%(体积分数)福尔马林缓冲液进行固定.吸取5 μL固定完毕的菌液,滴在无自发荧光的载玻片中央,加入甘油封片剂,盖上盖玻片后使用透明指甲油封闭.制成样本后用激光共聚焦显微镜观察结果(激发波长488nm,发射波长520nm,100倍油镜).
1.3.2 TOMA对脂肪酶的敏感性验证
酶处理条件:将0.238 g/L TOMA溶液5 mL(2.5μmoL),于 40℃水浴中预热 2min,脂肪酶用量0.5 ~3.5U,充分混匀后,于40℃、150 r/min 黑暗下处理20 min.滤膜过滤,滤液用于高效液相色谱(HPLC)分析检测水解后产物(避光).HPLC条件:色谱柱采用 Zorbax SB-C18柱(4.6 mm×250 mm,5μm);流动相为水∶乙腈 =60∶40(体积比)的磷酸盐缓冲液(0.02 mol/L,pH 值 6.99 ~ 7.01);柱温30℃;流量1mL/min;进样量10 μL;采用 PDA检测器,激发波长488nm,发射波长520nm.对照组中,使用等浓度未经酶解的TOMA溶液代替酶解液.结果以降解率表示,计算公式如下:
降解率=(酶解前TOMA含量-酶解后TOMA含量)/对照组TOMA含量×100%.其中,TOMA的含量以mg/mL为单位计.
1.3.3 TOMA对DNA扩增的抑制作用验证
分别取 500 μL金黄色葡萄球菌、大肠杆菌O157:H7、单增李斯特菌菌液于1.5 mL离心管中,6000r/min离心3 min,收集菌体,等体积无菌水重悬.按照细菌基因组DNA快速提取试剂盒操作说明提取基因组DNA.分别向提取的3种细菌DNA中加入 TOMA 溶液使终浓度达到 0.0、0.5、1.0、3.0、5.0、10.0、15.0、20.0 mg/L,充分混匀后于黑暗处静置5min,然后侧放于碎冰上,在卤钨灯下方约20cm处光照5min,使TOMA与DNA交联,同时钝化溶液中游离的TOMA分子.将经过光照处理的DNA溶液作为实时荧光定量PCR反应模板,考察TOMA对DNA扩增的抑制作用.用DNAStar Seqman模块(美国DNASTAR公司)对上述3种细菌分别进行基因序列比对、用Primer Express 2.0(美国应用生物系统公司)设计引物和探针.引物和探针序列如表1所示.

表1 引物和探针序列Table 1 Nucleotide sequences of primers and fluorogenic probes
荧光定量PCR反应体系总体积为25 μL,其中含10 ×Buffer 2.5μL、25mmol/L 的Mg2+溶液3.5μL、25mmol/L 的 dNTPs 1 μL、15 μmol/L 的前后引物各1μL、10 μmol/L 的探针 1 μL、模板溶液 2 μL、Taq DNA 聚合酶2.5U、DEPC 水12.5μL.反应条件:95℃预变性2min,95℃变性5s,然后降温至60℃并保持40s(收集荧光信号),该过程进行40个循环.反应结束后40℃保温2min.每个荧光定量PCR反应各进行3次平行实验,计算平均Ct值(荧光信号达到设定的阈值所经历的循环数)和样本标准差SD值.
2 结果与讨论
2.1 TOMA分子表征与预期特性
PMA对DNA扩增的抑制作用源于分子上连接的叠氮基团,其在光作用下于原位与DNA发生共价结合,使DNA在PCR反应中无法进行复制,有效性早已得到广泛承认[9-12].PMA分子完全不具细胞膜穿透性,但对一些具有可逆性的部分膜损伤细胞(如存在于环境样本中的细菌)以及部分膜完整的活菌,染料仍会穿透细胞膜,造成假阴性检测结果[13-15].此外,对经过紫外照射、冷冻灭菌等方法灭菌后仍保持细胞膜完整性的死菌,采用基于PMA的活菌PCR方法来评估会造成活细胞数目的严重高估,容易对检测结果产生误导[16].文中设计合成的TOMA是在总结PMA作用原理的基础上,利用噻唑橙类菁染料具有细胞膜通透性的特点,将PMA的功能与噻唑橙染料的优点进行联合应用.噻唑橙类染料能够自由渗透细胞膜,本身无自发荧光,与核酸结合后荧光显著增强.目前对这类染料的应用主要集中在核酸荧光标记定位方面.Carreon等[17]合成了4种结构不一的菁染料,对细胞线粒体进行了成像与定位.与已知的噻唑橙染料的结构相比,TOMA在喹啉环的一侧通过带有单个酯键的柔性碳链连接了叠氮基团,其相对分子质量为474.6,吸收光谱在488nm,发射光谱在520nm,分子结构经高分辨质谱仪及核磁共振仪表征无误.在分子设计上,TOMA分子柔性碳链上的酯键更靠近叠氮基团(而非噻唑橙基团),目的是使成功被水解脱落的叠氮基团分子更小,更有利于减小叠氮基团脱落后与DNA发生交联的概率.
TOMA的预期特性有3个:一是能自由穿透细胞膜;二是分子中的羧酸酯键能被生物体内大量存在的酯酶水解;三是未经酯酶水解的TOMA分子与DNA结合并发生交联后,能抑制DNA在PCR反应中的扩增.
2.2 TOMA对细菌的透膜效果
用Laica SP5激光共聚焦显微镜(100倍油镜,放大1000倍)观察TOMA穿透细胞膜与细菌DNA结合的效果.结果显示TOMA对3种细菌均具有良好的细胞膜穿透性,菌体经TOMA处理后发出强烈荧光.使用TOMA对活的大肠杆菌O157:H7处理后的显微图像及未经TOMA处理的大肠杆菌O157:H7的显微图像如图2所示.
从图2可见,菌悬液经过TOMA处理后,TOMA透过细胞膜进入菌体,与DNA发生特异性结合,所有菌体均产生了强烈绿色荧光,说明连接在TOMA分子上的叠氮基团并没有影响分子与DNA的结合以及荧光增强,TOMA与DNA结合后产生的荧光强度也没有因为叠氮基团的存在而出现明显下降.未经TOMA处理的菌体未检测出任何荧光.
噻唑橙类染料表现出与DNA的高结合率及结合后荧光强度的显著增长,使其很适合作为DNA探针[18].TOMA利用了噻唑橙染料可在细胞中自由扩散、通过被动运输透过细胞膜的特性,因此对活细胞和死细胞的DNA和RNA都可以进行结合染色(发出荧光),对革兰氏阳性和阴性细菌的核酸亦均可结合染色.TOMA自身没有荧光,是由于次甲基桥链两端的苯并噻唑(头部)和喹啉环(尾部)之间的自由旋转造成的,当有核酸参与时,头部与核酸的小沟结合,尾部插入碱基中,迫使头部和尾部发色团位置固定,导致荧光强度大大增强[19].因此,通过显微镜观察到,使用TOMA处理后细菌的菌体发出荧光,这可作为判断TOMA成功结合菌体DNA的标准.实验结果表明,TOMA分子具有自由穿透细菌壁膜的特性,能够成功穿透所有菌体细胞进入菌体内与DNA结合.TOMA对细胞膜的高穿透性,是利用TOMA建立起一套比PMA-qPCR检测方法更准确的活菌检测技术的前提.
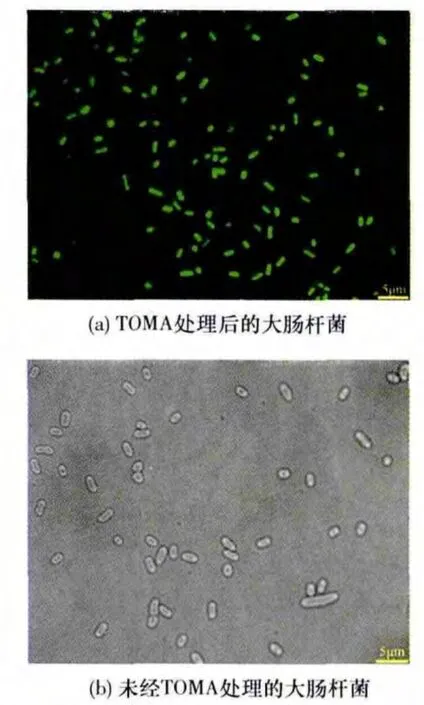
图2 经过TOMA处理和未经TOMA处理的大肠杆菌O157:H7的荧光显微图像Fig.2 Fluorescence images of Escherichia coli O157:H7 with and without TOMA treatment
2.3 TOMA的酶活敏感性
TOMA采用带有对酶活敏感的羧酸酯键的柔性碳链作为噻唑橙基团与叠氮基团的连接臂,是因为酯酶在细胞内普遍大量存在而且种类繁多.脂肪酶属于酯酶下的一个亚类,基本上存在于所有的生物体中[20],对维持生物体正常的功能运转有重要作用.目前已有成熟的基于活细胞酯酶活性的荧光染色试剂盒上市.如PromoKine公司的Live/Dead Cell Staining试剂盒,采用中性非荧光底物CTOMAein-AM对细胞进行处理,CTOMAein-AM渗透入活性细胞内后,分子的内酯键(羧酸酯键)被非特异性酯酶切断,生成不能穿过细胞膜的绿色极性荧光物质而被阻留在细胞体内[21].
在5mL 0.238g/L TOMA溶液中添加不同量的固定化脂肪酶,40℃、150r/min下处理20min后,使用HPLC上机检测.TOMA降解率随脂肪酶用量(以每μmol TOMA中添加的脂肪酶的量计)的变化情况如图3所示.
由图3可见,酶用量在0.2 ~1.0 U之间时,TOMA的降解率随酶用量增加呈上升趋势,降解率与酶用量之间成简单线性关系;继续增加酶用量至1.2、1.4 U,降解率基本不发生变化,说明过量的脂肪酶并不能提高TOMA的降解效果;而酶量不足会直接影响TOMA的降解效率,这一点与TOMA分子设计的初衷相符合.因为当细菌丧失生物代谢能力时,TOMA在菌体内将无法被降解,在后续的光照中叠氮基团在原位与DNA进行交联,从而抑制DNA在PCR反应中扩增.反之,在具有代谢活性的细胞中,TOMA中连接噻唑橙基团和叠氮基团的羧酸酯键被水解,叠氮基团从分子上脱落,失去后续与DNA交联的能力,对DNA扩增无影响.对于VBNC菌,菌体的酶活性可能较低,适当延长细菌与TOMA的孵育时间有助于反应完全.此外,从图3可以看出,本实验使用的脂肪酶最佳酶用量应为1.0U,此时TOMA的降解率达到83%以上.
体外实验结果表明,TOMA分子中的酯键设计达到了预期目的.实验中TOMA分子上连接的酯键对酯酶活性表现敏感,可在短时间内被脂肪酶水解,使叠氮基团脱落.而酯酶在活细胞内大量存在,因此,利用TOMA判断细胞是否具有生物代谢活性是可行的.设计TOMA分子,是希望通过细胞生物代谢活性的有无,选择性地抑制无代谢活性的细菌的DNA扩增,从而达到对活菌进行定量检测的目的.
然而,不同属不同株的细菌体内表达的酶类与酶量之间有差异,不同存活状态下细菌代谢活力也不尽相同.因此,对食源性致病菌体内酯酶种类、活性进行鉴定和测定后,探索几种典型食源性致病菌在不同存活状态下酯酶代谢活性水平的差异特征,从而进一步优化TOMA的使用方法,是下一步研究的方向.
2.4 TOMA对DNA扩增的抑制效果
TOMA分子中的叠氮基团与EMA/PMA中的叠氮基团相同.当分子在水溶液状态下被光照时,叠氮基团迅速失去自由氮产生活性氮烯,与水形成羟胺衍生物,当分子处于与DNA结合状态而非游离状态时,由于氮烯对DNA的进攻,分子的叠氮基团与DNA形成共价键,从而在原位生成共价复合物[22].与叠氮基团共价结合后的DNA无法参与PCR扩增.
TOMA浓度对DNA扩增的影响如图4所示.由图4可见,3种细菌的DNA在PCR反应中的Ct值均随着TOMA浓度的增大而显著增大,表明TOMA与DNA结合后,分子上的叠氮基团对DNA扩增的抑制效果明显,并且抑制效果与TOMA浓度呈正相关.使用低浓度(0.5、1.0 mg/L)TOMA 处理的 DNA溶液,其PCR反应后的Ct值与TOMA浓度为0的DNA溶液的相比增幅不大,说明使用过低浓度的TOMA对DNA扩增的影响不大,因此,采用低浓度的TOMA在活菌检测中可能会造成假阳性的结果.实验中TOMA浓度为10.0 mg/L时,TOMA对DNA的抑制作用明显,此时Ct值达到36左右,继续增大TOMA浓度至15.0 mg/L对Ct值仅有轻微影响.而当TOMA浓度达到20.0mg/L时,整个PCR反应过程无检出,推测可能过高浓度的TOMA会对PCR反应有抑制作用.实验表明,TOMA分子中连接的叠氮基团在光照下能成功对DNA进行共价交联作用,在合适的浓度条件下能充分抑制DNA在PCR反应中的扩增.
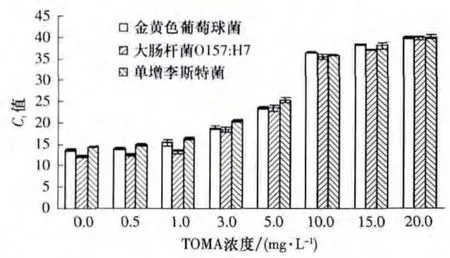
图4 TOMA浓度对DNA扩增的影响Fig.4 Effect of TOMA concentration on DNA amplification
3 结语
文中设计合成了一种新型噻唑橙衍生物TOMA,并分别从3个方面对TOMA的预期特性进行验证.结果表明:连接叠氮基团的TOMA分子可以自由透过细菌的细胞膜;TOMA分子上的酯键能在酶作用下被快速高效降解;TOMA分子对DNA扩增具有明显的抑制作用.TOMA分子的预期功能特性在体外实验中得到了初步证实,为进一步运用TOMA对细菌进行活菌检测研究提供了重要参考.后续研究中,拟将TOMA与实时荧光PCR技术结合起来,利用死/活细菌体内生物活性的差异来对致病菌进行快速有效的检测.由于TOMA对细菌体内的生物活性敏感度高,TOMA应用在致病菌活菌检测技术中能同时克服传统qPCR法假阴性和PMA-qPCR法假阳性的缺点,有效识别VBNC菌、非膜损伤死菌等,为致病菌活菌检测手段提供新的方向.
[1]Riedy M C,Muirhead K A,Jensen C P,et al.Use of a photolabeling technique to identify nonviable cells in fixed homologous or heterologous cell populations[J].Cytometry,1991,12(2):133-139.
[2]Marouani-Gadri N,Firmesse O,Chassaing D,et al.Potential of Escherichia coli O157:H7 to persist and form viable but non-culturable cells on a food-contact surface subjected to cycles of soiling and chemical treatment[J].International Journal of Food Microbiology,2010,144(1):96-103.
[3]Nogva H K,Dromtorp S M,Nissen H,et al.Ethidium monoazide for DNA-based differentiation of viable and dead bacteria by 5'-nuclease PCR [J].BioTechniques,2003,34(4):804-813.
[4]Rudi K,Moen B,Drømtorp S M,et al.Use of ethidium monoazide and PCR in combination for quantification of viable and dead cells in complex samples[J].Applied and Environmental Microbiology,2005,71(2):1018-1024.
[5]Nocker A,Sossa-Fernandez P,Burr M D,et al.Use of propidium monoazide for live/dead distinction in microbial ecology [J].Applied and Environmental Microbiology,2007,73(16):5111-5117.
[6]Pisz J M,Lawrence J R,Schafer A N,et al.Differentiation of genes extracted from non-viable versus viable micro-organisms in environmental samples using ethidium monoazide bromide[J].Journal of Microbiological Methods,2007,71(3):312-318.
[7]Varma M,Field R,Stinson M,et al.Quantitative real-time PCR analysis of total and propidium monoazide-resistant fecal indicator bacteria in wastewater[J].Water Research,2009,43(19):4790-4801.
[8]Wagner A O,Malin C,Knapp B A,et al.Removal of free extracellular DNA from environmental samples by ethidium monoazide and propidium monoazide[J].Applied and Environmental Microbiology,2008,74(8):2537-2539.
[9]Agustí G,Codony F,Fittipaldi M,et al.Viability determination of Helicobacter pylori using propidium monoazide quantitative PCR [J].Helicobacter,2010,15(5):473-476.
[10]Rawsthorne H,Dock C N,Jaykus L A.PCR-based method using propidium monoazide to distinguish viable from nonviable Bacillus subtilis spores[J].Applied and Environmental Microbiology,2009,75(9):2936-2939.
[11]Vesper S,McKinstry C,Hartmann C,et al.Quantifying fungal viability in air and water samples using quantitative PCR after treatment with propidium monoazide(PMA)[J].Journal of Microbiological Methods,2008,72(2):180-184.
[12]Andorrà I,Esteve-Zarzoso B,Guillamón J M,et al.Determination of viable wine yeast using DNA binding dyes and quantitative PCR [J].International Journal of Food Microbiology,2010,144(2):257-262.
[13]Fittipaldi M,Nocker A,Codony F.Progress in understanding preferential detection of live cells using viability dyes in combination with DNA amplification[J].Journal of Microbiological Methods,2012,91(2):276-289.
[14]Kralik P,Nocker A,Pavlik I.Mycobacterium avium subsp paratuberculosis viability determination using F57 quantitative PCR in combination with propidium monoazide treatment[J].International Journal of Food Microbiology,2010,141(Suppl 1):S80-S86.
[15]Liang N,Dong J,Luo L,et al.Detection of viable Salmonella in lettuce by propidium monoazide real-time PCR[J].Journal of Food Science,2011,76(4):M234-M237.
[16]Nocker A,Sossa K E,Camper A K.Molecular monitoring of disinfection efficacy using propidium monoazide in combination with quantitative PCR [J].Journal of Microbiological Methods,2007,70(2):252-260.
[17]Carreon J R,Stewart K M,Mahon Jr K P,et al.Cyanine dye conjugates as probes for live cell imaging [J].Bioorganic & Medicinal Chemistry Letters,2007,17(18):5182-5185.
[18]Isacsson J,Westman G.Solid-phase synthesis of asymmetric cyanine dyes[J].Tetrahedron Letters,2001,42(18):3207-3210
[19]陈秀英,牛艳明,郭琳,等.噻唑橙核酸荧光探针的合成及光谱性质研究[J].化学研究与应用,2010,22(10):1267-1271.Chen Xiu-ying,Niu Yan-ming,Guo Lin,et al.Synthesis and spectral properties of thiazole orange compounds as DNA fluorescent probes[J].Chemical Research and Application,2010,22(10):1267-1271.
[20]Svendsen A.Lipase protein engineering[J].Biochimica et Biophysica Acta(BBA)-Protein Structure and Molecular Enzymology,2000,1543(2):223-238.
[21]Grieshaber P,Lagrèze W A,Noack C,et al.Staining of fluorogold-prelabeled retinal ganglion cells with cTOMAein-AM:a new method for assessing cell vitality[J].Journal of Neuroscience Methods,2010,192(2):233-239.
[22]Coffman G L,Gaubatz J W,Yielding K L,et al.Demonstration of specific high affinity binding sites in plasmid DNA by photoaffinity labeling with an ethidium analog[J].Journal of Biological Chemistry,1982,257(22):13205-13207.
猜你喜欢
云南化工(2021年7期)2021-12-21 07:27:22
原子与分子物理学报(2021年1期)2021-03-29 07:28:26
商品与质量(2019年32期)2019-11-29 05:56:00
天然产物研究与开发(2018年8期)2018-09-10 05:48:24
天然产物研究与开发(2018年4期)2018-05-07 06:47:45
火工品(2018年1期)2018-05-03 02:27:56
中国资源综合利用(2017年3期)2018-01-22 02:45:40
合成化学(2015年9期)2016-01-17 08:57:14
郑州大学学报(工学版)(2015年1期)2015-03-24 00:55:36
中成药(2014年9期)2014-02-28 22:28:55
