
硫化型催化剂的活性相及其加氢脱硫活性
2013-05-03 01:54:06徐黎明高玉兰彭绍忠
石油化工 2013年3期
徐黎明,高玉兰,彭绍忠
(中国石化 抚顺石油化工研究院,辽宁 抚顺 113001)
近年来世界范围内原油劣质化倾向日益明显,石油馏分中硫、氮和芳烃的含量日益提高,与此同时各国对环保的要求却越来越高。加氢技术是生产清洁油品的有效手段之一,其中加氢催化剂是加氢技术的关键[1]。常规的加氢催化剂成品中金属组分以氧化态形式存在,而实际使用时真正起活性作用的是金属硫化物,因此加氢催化剂在使用前需将氧化态金属组分转化为硫化态。目前工业上广泛应用的预硫化方式有两种:器内预硫化和器外预硫化[2-5]。工业应用结果表明,与器内预硫化相比,器外预硫化技术具有开工时间短、开工步骤简单、炼厂设备投资少、对人体危害小以及对环境污染小等优点。但器外预硫化与器内预硫化一样,催化剂需经氧化态进行预硫化,造成生产步骤繁琐、制备工艺复杂、耗能耗时、经济性较差等问题;而且由于金属氧化物与载体的相互作用过强,使金属组分的硫化不完全,不能充分发挥作用,造成金属利用率低。为了解决这些问题,直接硫化技术应运而生。
直接硫化型加氢催化剂(简称硫化型催化剂)中活性金属直接以硫化态形式存在,不用经氧化态这一中间过程,不仅简化了催化剂生产步骤、降低了生产成本,而且由于金属硫化度的提高,最终提高了催化剂的活性。文献[6]公开了一种改性钴钼基硫化型催化剂的制备方法,通过配制硫代钼酸铵溶液,共沉淀钼、钴和第三种过渡金属组分,在氮气保护下焙烧,制得黑色粉末状催化剂。文献[7]主要报道了采用可溶性硫代钼酸盐和硫代钨酸盐将钼和钨的前体引入到常规的催化剂载体中的方法,在氮气保护下焙烧,再用含镍和钴的溶液浸渍,然后在氮气保护下焙烧,从而制得含钨、钼、镍和钴的负载型硫化型催化剂。该催化剂的加氢活性好,目前尚处于实验室探索阶段。总体来说,对硫化型催化剂的制备研究还不够深入,对金属硫化物形成机理的研究更是鲜有报道。
本工作制备了镍钼基硫化型催化剂,通过对硫化过程的分析和表征,初步探索了硫化型催化剂活性相的形成过程,并对其加氢脱硫活性进行了评价。
1 实验部分
1.1 催化剂的制备
以常规的钼酸铵及硫化铵为原料,在碱性条件下煮沸合成金属硫化物前体(NH4)2MoS4[8],将其与硝酸镍及有机添加剂配成所需浓度的浸渍液,用该浸渍液浸渍氧化铝载体,将浸渍后的载体在120 ℃下干燥3 h,即得硫化型催化剂C-2和C-4。C-2和C-4催化剂的制备方法相同,但金属配比及含量不同[9]。 为考察有机添加剂的作用,采用无添加剂的浸渍液制备了催化剂C-3,C-3与C-2催化剂的制备条件相同,只是浸渍液中未加入有机添加剂。为了与硫化型催化剂对比,又分别制备了氧化态催化剂C-1和器外预硫化催化剂A-1。将氧化钼、碱式碳酸镍和磷酸煮沸制成钼镍磷溶液,用其浸渍氧化铝载体,在100 ℃下干燥3 h,500 ℃下焙烧3 h,即得氧化态催化剂C-1[10]。将C-1催化剂按文献[11]报道的方法制成器外预硫化催化剂A-1。催化剂的组成见表1。

表1 催化剂的组成Table 1 Compositions of prepared catalysts
1.2 分析与表征
采用热电公司Multilab 2000型X射线光电子能谱仪进行XPS表征,激发源Mg Kα,分析室真空度高于10-6Pa,以C1s(284.6 ev)为内标校正核电效应。使用XPSPEAK41软件进行XPS谱图的拟合,假定各单一物种峰形为高斯形,根据手册和文献中不同钼、硫和镍价态的结合能和半峰宽,以其相对含量为变量进行谱图拟合,分析钼、硫和镍组分的不同形式及其含量。
采用日本电子公司JEM-2100型透射电子显微镜观察试样的表面形貌,点分辨率0.23 nm,线分辨率0.14 nm。
采用Netzsch公司DSC204HP型示差扫描量热分析仪进行高压DSC分析,体系内为高压H2氛围,H2压力为3.5 MPa,H2流量为20 mL/min,升温速率为10 ℃/min,催化剂的装填量为30 mg,升温范围从室温至400 ℃。
1.3 催化剂的评价
催化剂的评价在自建的10 mL微型反应器上进行。原料油为常二线油,硫含量为6 524.1 μg/g,氮含量为50.6 μg/g。氧化态催化剂C-1先用含硫煤油进行器内预硫化,切换原料油恒温稳定2 h后,每隔1 h取样,进行硫氮含量的分析。评价硫化型催化剂C-2,C-3,C-4时,直接通入原料油和H2,升温至反应温度后恒温稳定2 h,每隔1 h取样,进行硫氮含量分析。A-1的评价方法与硫化型催化剂相同。评价催化剂的工艺条件相同:反应温度350 ℃,反应压力3.4 MPa,氢油体积比360,原料油的液态空速2.0 h-1。
2 结果与讨论
2.1 硫化型催化剂的高压DSC分析结果
催化剂上金属的硫化过程就是活性相的形成过程。通过高压DSC模拟硫化型催化剂C-2的硫化过程,分析硫化过程中发生的反应,并与器外预硫化催化剂A-1进行比较,结果如图1所示。由图1可看出,硫化型催化剂C-2的DSC曲线在140 ℃左右有一个大的吸热峰,归属于催化剂的脱水;215,282,367 ℃附近的3个放热峰为硫化放热峰。为进一步说明硫化过程中所发生的反应,对这3个放热峰温度区间的试样进行XPS分析,获得催化剂表面各物种的分析数据,用拟合软件对XPS谱图进行拟合,由拟合峰面积计算催化剂表面钼的含量。计算结果表明,硫化物前体先由+6价变为+5价,再逐渐转变为+4价(即二硫化钼)。前体由+6价变为+5价是较为容易的过程,因此转变温度低且转化完全,随温度的进一步提高,逐步转化生成+4价二硫化钼。温度升至400 ℃时仍有少量+5价前体未转化完全,这是由DSC实验升温速率快导致的,在此温度下恒温1 h则前体完全转化为二硫化钼。由图1还可看出,与器外预硫化催化剂A-1的集中放热峰相比,硫化型催化剂C-2的放热量明显降低且分散,硫化型催化剂的第一个放热峰温度降低,但峰值很小,峰顶与基线基本相平。众所周知,催化剂在开工时的集中放热是制备硫化型催化剂必须解决的重要问题之一[12],放热量大和放热集中都可能导致开工时催化剂床层飞温,损伤催化剂,以至造成安全事故。硫化型催化剂的放热量小且分散,可保证催化剂的正常开工使用。

图1 A-1和C-2催化剂的高压DSC曲线Fig.1 High pressure DSC curves of the A-1 and C-2 catalysts.
2.2 有机助剂对硫化型催化剂放热效应的影响
由于硫化型催化剂的特殊性,在催化剂制备过程中加入有机助剂对催化剂上活性金属存在状态有很大的影响。通过对无有机助剂的催化剂C-3与添加有机助剂的催化剂C-2进行高压DSC和XPS表征来揭示有机助剂在活性相形成过程中的作用,高压DSC分析结果见图2。由图2可知,有机助剂的加入从根本上改变了催化剂的放热曲线,即改变了催化剂上金属的硫化过程,形成了不同的活性相。C-2催化剂表面的金属镍是与钼一起硫化的,分散在二硫化钼的表面,形成了高活性的Ni—Mo—S活性相,未出现金属镍的放热峰。C-3催化剂的两个放热峰一个是生成二硫化钼的放热峰,一个是生成硫化镍产生的放热峰。这说明未加入有机助剂时,金属镍未与钼形成Ni—Mo—S活性相,而是形成了低活性的二硫化钼和硫化镍相[13]。因此制备过程中引入有机助剂可促进活性金属形成高活性的二类活性相,提高催化剂的加氢活性,并使硫化型催化剂的放热峰弥散变小,可避免因活化放热量过大、过于集中而产生飞温。

图2 C-2和C-3催化剂的高压DSC曲线Fig.2 High pressure DSC curves of the C-2 and C-3 catalysts.
2.3 硫化型催化剂的TEM分析结果
根据Co—Mo—S模型,如果载体与金属之间的相互作用过强,则生成低活性的活性相,在载体表面上呈单层分布;如果载体与金属的相互作用较弱,金属易硫化,易形成高活性的活性相,在载体表面呈多层分布,活性中心为金属叠层的边角棱位。计算二硫化钼片晶的平均片层长度(L,nm)[14]和平均堆叠层数(N)[15],可更加直观地比较活性相的变化。平均片层长度和平均堆叠层数的计算公式如下:
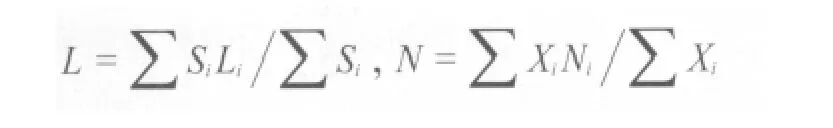
式中,Si为片层长度为Li的片层数目;Xi为具有Ni层的片晶个数。二硫化钼片晶的片层长度增大,片晶层数增多,在载体表面形成较大的活性堆垛,这对于噻吩和喹啉等反应物克服空间位阻吸附在活性中心位上非常有利。但堆叠层数和平均长度也并非越大越好,片晶的堆叠层数和平均长度增加,一方面可以生成高活性的二类活性相,但另一方面二硫化钼片晶易团聚,导致活性金属组分的分散度降低,使反应分子可接近的活性中心总数减少。因此,适宜的堆叠层数和稍短的平均长度对提高催化剂的加氢活性有利。一般认为,堆叠层数为3~5最好。
对评价后的催化剂进行TEM表征,表征结果见图3。每个试样随机选取20张TEM照片,用作图软件统计照片中二硫化钼片晶的平均堆叠层数和平均片层长度,结果见表2。由表2可看出,C-1催化剂的二硫化钼片晶大多为1或2层,说明载体与活性金属的相互作用较强,金属难以硫化,形成了低活性的活性相。A-1催化剂的二硫化钼片晶的堆叠层数大于C-1催化剂,这与文献[12]报道的器外预硫化催化剂更易硫化的结论相吻合;而C-2催化剂的二硫化钼片晶的堆叠层数较多,片晶的长度较长,形成了高活性的Ni—Mo—S相,载体与活性金属的相互作用小,这与载体只干燥不焙烧,载体与金属的相互作用属于范德华力有关。

图3 评价后催化剂的TEM照片Fig.3 TEM images of the used catalysts.

表2 二硫化钼片晶的平均堆叠层数和平均长度Table 2 Average layer number and length of MoS2 lamellar crystals
2.4 硫化型催化剂的加氢活性
催化剂的评价结果见表3。

表3 催化剂的评价结果Table 3 Evaluation results of the catalysts
由表3可见,在硫化型催化剂C-2和C-4的金属含量低于氧化态催化剂C-1的情况下,其加氢脱硫活性均优于C-1催化剂,这是因为硫化型催化剂表面的钼完全以二硫化钼形式存在,钼组分完全硫化,镍组分高度分散在二硫化钼的边角等位置,与二硫化钼形成高活性的Ni—Mo—S相,活性金属的有效利用率高;活性金属和硫组分在载体上均匀分布也是C-2和C-4催化剂活性高的原因之一。与器外预硫化催化剂A-1相比,C-2和C-4催化剂的加氢脱硫活性优于A-1催化剂。在总金属含量比C-1催化剂降低30%的情况下,C-4催化剂的活性仍较高,说明相对于氧化态催化剂,硫化型催化剂由于采用了新型的制备方法,适当降低催化剂上的总金属含量也可达到氧化态催化剂的脱硫水平。同时,适当减小金属配比对硫化型催化剂的活性基本没有影响,这说明新的制备方法提高了金属镍在载体表面的分散度。
3 结论
1)硫化过程中硫化型催化剂上金属先经+5价的过渡态,再逐步生成二硫化钼相。有机助剂的加入不仅改变了催化剂的硫化过程,促进了金属镍在二硫化钼表面的分布,形成高活性的Ni—Mo—S相,还使硫化时的放热峰弥散减小,可避免开工时催化剂床层飞温。
2)由于采用了新型的制备技术,硫化型催化剂表面上活性金属完全硫化,形成高活性的Ni—Mo—S相,催化剂的加氢脱硫活性高。
[1]李大东.加氢处理工艺与工程[M].北京:中国石化出版社,2004:4-84.
[2]刘畅,祁兴国,马守波,等.加氢催化剂器外预硫化技术进展[J].化学工程师,2006(1):33-38.
[3]王月霞.加氢催化剂的器外预硫化[J].炼油设计,2000,30(7):57-58.
[4]Europeene de Retraitement de Catalyseurs Eurecat.Process for Presulfurizing with Phosphorous and/or Halogen Additive:US,4983559[P].1991-01-08.
[5]CRI International.Method of Presulfurizing a Hydrotreating,Hydrocracking or Tail Gas Treating Catalyst:US,5688736[P].1997-11-18.
[6]华东师范大学.一种改性钴钼基硫化物催化剂及其制备方法:中国,1569331[P].2005-01-26.
[7]中国石油天然气集团公司,中国石油大学(华东).一种硫化型加氢催化剂及其制备方法:中国,1557917[P].2004-12-29.
[8]柴永明,赵会吉,柳云骐,等.四硫代钼酸铵制备方法改进[J].无机盐工业,2007(5):21-51.
[9]中国石油化工股份有限公司,中国石油化工股份有限公司抚顺石油化工研究院.硫化型催化剂的制备方法:中国,101722055[P].2010-06-09.
[10]杨占林,彭绍忠,刘雪玲,等.P改性对Mo-Ni/γ-Al2O3催化剂结构和性质的影响[J].石油化工,2007,36(8):784-788.
[11]中国石油化工股份有限公司,中国石油化工股份有限公司抚顺石油化工研究院.一种加氢催化剂应用前的处理方法:中国,101417230[P].2009-04-29.
[12]高玉兰,方向晨.加氢催化剂器外预硫化技术的研究[J].石油炼制与化工,2005,36(7):1-3.
[13]徐黎明,高玉兰,曹凤兰,等.硫化型加氢催化剂的制备研究[J].石油炼制与化工,2009,40(4):1-4.
[14]Ferdous D,Dalai A K,Adjaye J,et al.Surface Morphology of NiMo/Al2O3Catalysts Incorporated with Boron and Phosphorus:Experimental and Simulation[J].Appl Catal,A,2005,294(1):80-91.
[15]Hensen E J M,Kooyman P J,van der Meer Y,et al.The Relation Between Morphology and Hydrotreating Activity for Supported MoS2Particles[J].J Catal,2001,199(2):224-235.
猜你喜欢
安徽化工(2022年5期)2022-09-28 05:25:44
环境工程技术学报(2022年3期)2022-06-05 07:20:30
陶瓷学报(2021年4期)2021-10-14 08:57:36
电工技术学报(2021年12期)2021-07-01 05:42:42
表面工程与再制造(2019年1期)2019-05-11 08:51:46
天然产物研究与开发(2019年1期)2019-03-01 05:41:04
科学与技术(2018年8期)2018-04-26 06:50:56
中国铸造装备与技术(2015年5期)2015-12-10 10:23:39
中国塑料(2015年2期)2015-10-14 05:34:27
电源技术(2015年11期)2015-08-22 08:50:26
