
N-乙酰-L-色氨酸减轻H2 O2诱导小鼠海马神经元细胞凋亡
2013-03-22 05:58于树娜王志芳杜晓东姜红心史才兴蒋吉英
基础医学与临床 2013年4期
于树娜,王志芳,杜晓东,姜红心,李 进,史才兴,蒋吉英*
(1.潍坊医学院人体解剖学教研室,山东潍坊261053;2.即墨市人民医院神经内科,山东郎墨266200;3.潍坊医学院形态学实验室,山东潍坊261053)
脑梗死是现今人类病死率最高的3大疾病之一,脑缺血再灌注损伤是脑梗死最常见的病理表现。目前临床上对该病尚无特效疗法。近年来大量研究表明,脑缺血再灌注损伤后,神经元的死亡是通过细胞坏死和细胞凋亡两个途径进行的[1-2],其中细胞坏死是一个不可逆的过程,而细胞凋亡是可逆的过程,与梗死灶的发展扩大有关,可能决定了最终的梗死体积[3]。因此,阻断缺血神经元的凋亡,从而逆转神经元损伤,已成为缺血性脑血管病的研究热点。海马神经元对低氧极为敏感,具有较高的易损性,以往有研究表明,H2O2可引起神经细胞的氧化损伤[3]。因此,本研究用E15d的小鼠分离海马神经元(primary hippocampal neurons,PHN),用 H2O2诱导PHN细胞凋亡,建立脑缺血低氧损伤的细胞模型,观察 N-乙酰-L-色氨酸(N-acetyl-L-tryptophan,L-NAT)对PHN细胞凋亡的影响,为提高脑缺血再灌注损伤的疗效提供实验依据。
1 材料与方法
1.1 胚胎鼠PHN的培养、凋亡诱导及L-NAT干预
断头取E15d昆明小鼠脑,分离海马,0.125%胰蛋白酶消化,以1×106的细胞密度接种于经多聚赖氨酸包被的培养皿,加入神经基础培养液(含2%B27、2 mmol/L谷氨酰胺和100 U/mL青-链霉素),培养6 d后,将细胞分为对照组、H2O2组、H2O2+L-NAT组,每组2孔,待贴壁细胞的数量达80%时,用600μmol/L H2O2诱导细胞凋亡,L-NAT组在H2O2诱导前用30μmol/L L-NAT预处理2 h。18~20 h后,收集细胞或上清液进行相关的检测。所有的实验结果重复3次。
1.2 免疫荧光染色
4%多聚甲醛后固定、0.5%Triton X-100及5%BSA封闭,滴加多克隆抗 caspase-3抗体(1∶200,Cell Signaling公司),4℃孵育过夜;FITC-conjugated donkeny anti-rabbit IgG(1∶200,Jackson Immunoresearch 公司),4℃孵育60min。以PBS代替一抗作阴性对照。
1.3 细胞存活率检测
常规锥虫蓝染色,倒置相差显微镜下计数(200倍)计数20个视野蓝色的细胞数(即死亡细胞数)、非蓝色细胞数(即存活的细胞数),根据以下公式计算细胞存活率:细胞存活率(%)=存活的细胞数/(死亡细胞数+存活的细胞数)。
1.4 Rhodam ine 123染色
PHN细胞培养至对数生长期,吸弃培养基,加入终浓度为2μmol/L的 Rhodamine 123染色液。37℃孵育30 min;PBS冲洗10 min×3次,荧光显微镜下观察、拍照。
1.5 Caspase-3活性分析
取对数生长期的PHN细胞,参考文献[3],405 nm比色检测各组细胞中caspase-3的活性。
1.6 乳酸脱氢酶(Lactate dehydrogenase,LDH)活性检测
按照说明书检测不同实验组PHNs细胞培养上清液中所释放LDH的量。即收集各组细胞的上清液,离心,与 LDH反应复合物混合,避光反应30 min,用酶标仪测定492 nm的吸光度。
1.7 胞质蛋白与线粒体蛋白的分离
在培养的各组细胞中加入胞质提取缓冲液,冰浴上匀浆,4℃,2 000×g,离心5 min;收集上清液入一新的离心管,4℃,15 000×g,离心25 min;其上清液即为胞质蛋白。将其沉淀用RIPA缓冲液在冰浴上裂解10 min即为线粒体蛋白,分别用Western blot检测凋亡诱导因子(apoptosis-inducing factor,AIF)和细胞色素C(cytochrome c,Cytc)等线粒体促凋亡因子在胞质蛋白和线粒体蛋白中的表达。
1.8 W estern blot
常规提取总蛋白,用BCA法检测各组蛋白浓度,取50μg总蛋白进行聚丙烯凝胶电泳,转膜、5%脱脂奶粉封闭1 h(4℃);滴加多克隆抗caspase-3抗 体 (1∶600,Cell Signaling 公 司)、CytC 抗 体(1∶600,Cell Signaling 公司)、AIF 抗体(1∶600,Cell Signaling公司),4℃过夜;TBST洗膜15 min×4次;滴加二抗,室温2 h,TBST洗膜15 min×4次;加入ECL试剂X线曝光成像。
1.9 统计学分析
采用SPSS11.0统计软件对结果进行分析,结果用均数±标准差(±s)表示,用单因素方差分析(ANOVA)进行统计学分析。
2 结果
2.1 L-NAT对细胞形态的影响
原代培养的大鼠海马神经元在接种后1 h左右即可贴壁,培养2 h大部分细胞己经贴壁生长,此时可见少数神经元伸出1~2个突起;培养5~6 d时,神经元胞体呈椭圆形或三角形,胞体周围光晕明显,神经元突起的主干和分支明显延长、增粗,相互联系增多。H2O2诱导后,神经元细胞膜变得粗糙不光滑,部分细胞变圆,胞体肿胀,细胞失去原有形态,突起减少或消失。与H2O2组相比,L-NAT组神经元的胞体较完整,大部分细胞存在少量的突起,细胞突起存在少量的联系,突起的数量较正常组减少(图1)。

图1 L-NAT抑制H2O2诱导的形态改变Fig 1 The effect of L-NAT on H2 O2-induced morphological changes of PHNs(×400)
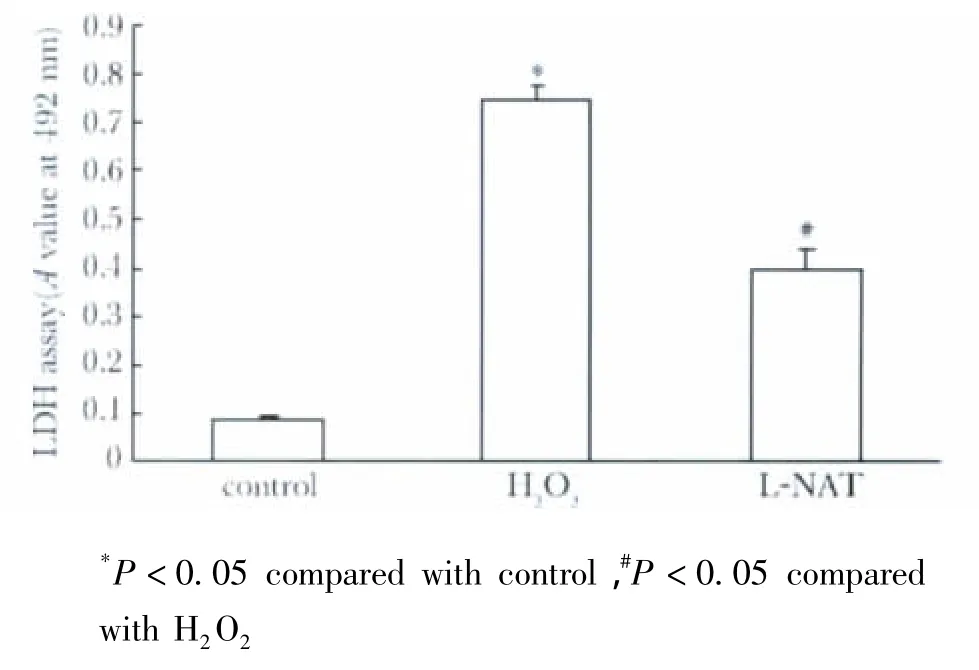
图2 L-NAT对H2 O2诱导的PHN细胞LDH的释放的影响Fig 2 The effect of L-NAT on H 2O2-induced LDH-release of PHNs
2.2 L-NAT对细胞活性的影响
本研究首先观察不同浓度L-NAT(0.001~200μmol/L)对PHNs细胞培养液中LDH浓度的影响,发现10和30μmol/L L-NAT对PHNs细胞均有保护作用,100μmol/L时即出现细胞毒性作用,其中30μmol/L L-NAT对细胞的保护作用更好,因此,本研究观察30μmol/L L-NAT对PHNs的保护作用。结果显示,L-NAT组细胞上清液中LDH的浓度明显低于H2O2组(P<0.05)(图2)。
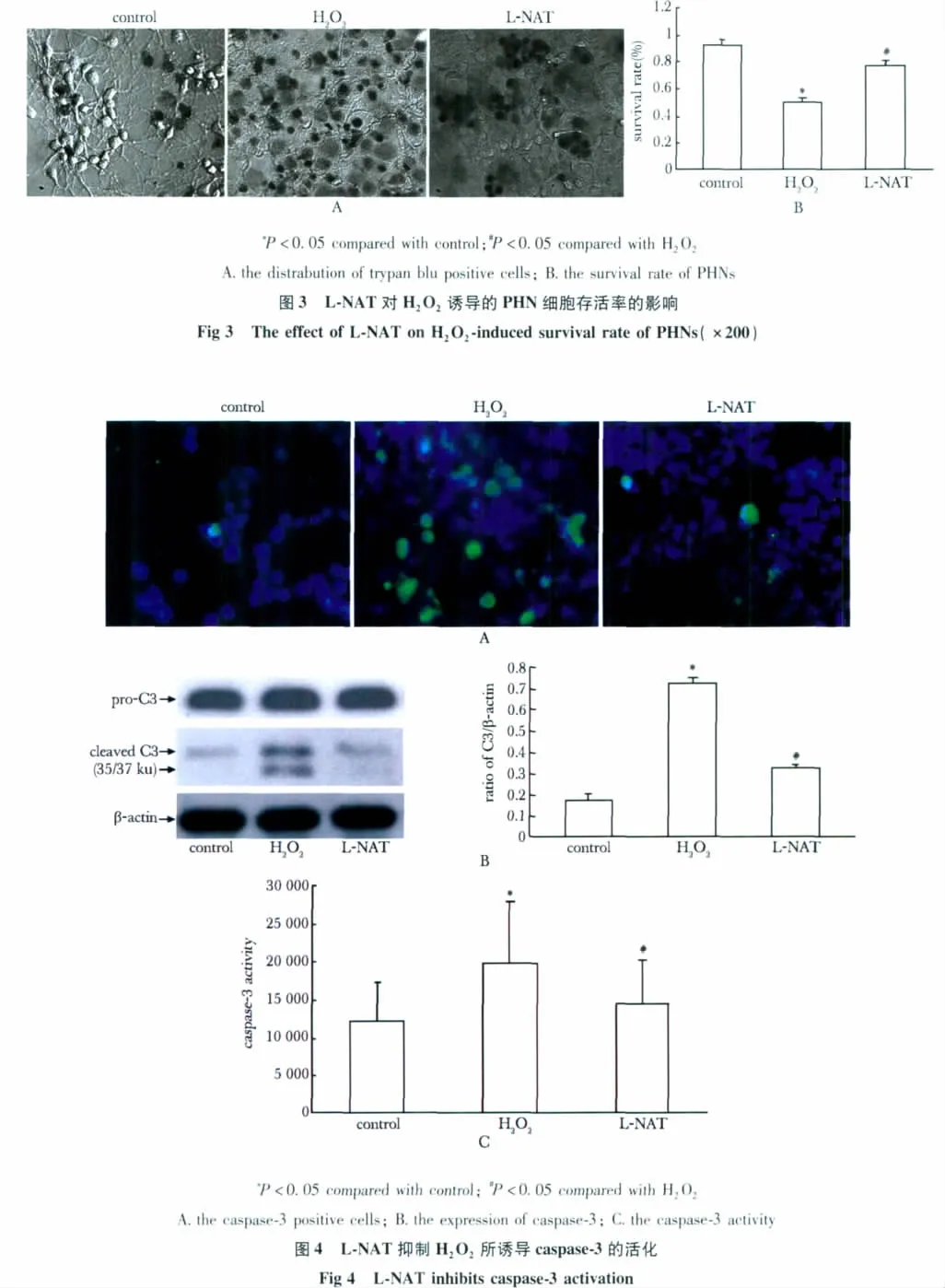
对照组可见少量的锥虫蓝阳性细胞,H2O2诱导后,出现大量的阳性细胞,L-NAT干预后,阳性细胞的数量降低(图3A)。H2O2可使细胞存活率降低,L-NAT可使之逆转(P<0.05)(图3B)。
2.3 L-NAT对caspase-3的影响
Caspase-3阳性细胞及蛋白在H2O2组的表达较L-NAT组高(P<0.05)(图4A,B);caspase-3的活性在H2O2诱导后升高,L-NAT可使之逆转(P<0.05)(图4C)。
2.4 L-NAT对线粒体膜势能(ΔΨm)的影响
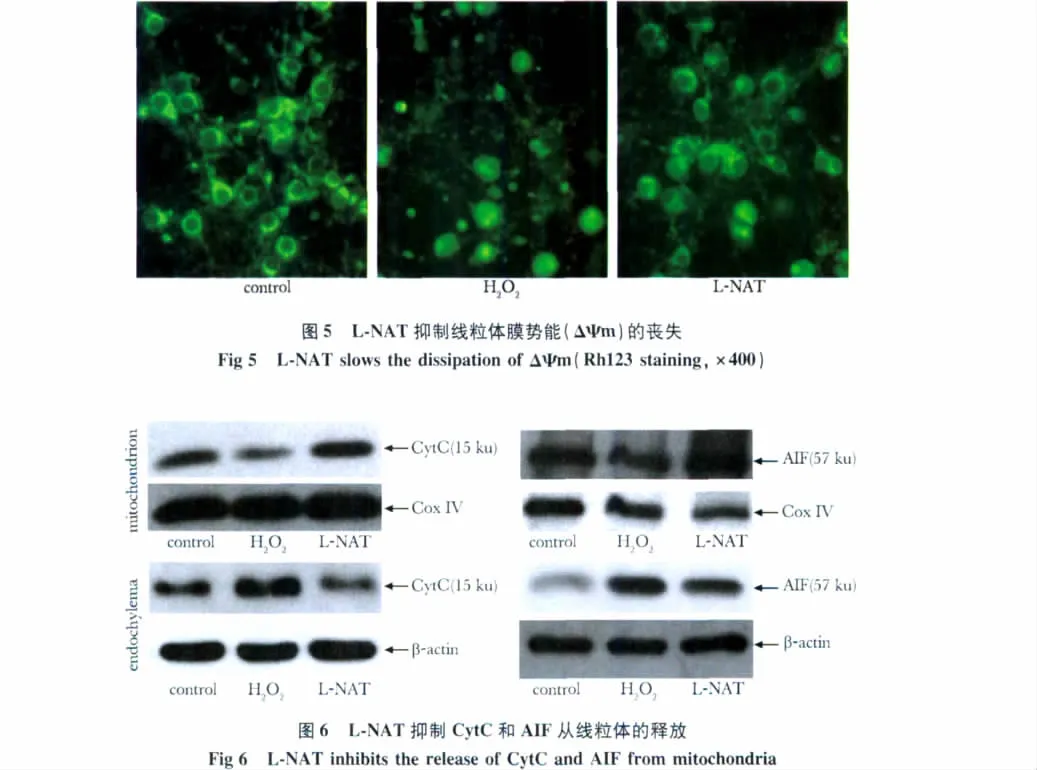
对照组Rh123阳性产物呈点状或颗粒状分布于胞质中,H2O2组阳性反应减弱,阳性产物呈弥散性分布;L-NAT可使部分细胞恢复颗粒状分布的特点(图5)。
2.5 L-NAT对线粒体促凋亡因子释放的影响
CytC和AIF在对照组线粒体中的表达较高;在H2O2组由于其向胞质的释放,使其在线粒体中的量降低,而在胞质中的表达增高;L-NAT可减轻这一改变(图6)。
3 讨论
缺血再灌注损伤的发病机制尚未明确,现有研究表明,线粒体对细胞凋亡的发生起着核心作用,可以看作是细胞凋亡的执行中心,因为几乎所有导致细胞凋亡的刺激因素都会诱发线粒体结构的破坏和功能的障碍[4-6]。线粒体内膜对离子通透性的严格限制对形成和维持正常的线粒体膜势能(ΔΨm)至关重要,当细胞受到某些刺激(如药物、射线、氧化剂、钙超载等)后线粒体外膜的通透性增高,内膜的电化学梯度减低,线粒体膜势能(ΔΨm)消失,CytC、AIF和Smac(second mitochondria-derived activator of caspase)等线粒体促凋亡因子释放[7]。CytC的释放可启动Apaf-1/caspase-9/caspase-3复合体的形成,激活caspase-3,从而形成caspases依赖性的细胞凋亡[8]。线粒体释放的AIF的胞核转位在急性神经元损伤中也发挥重要作用[9],与CytC的作用方式不同,AIF通过caspase非依赖性凋亡途径发挥作用[10]。有研究表明,新生鼠脑缺血再灌注损伤后,AIF即从线粒体释放,转移至胞核内,且其出现的时间较CytC早[11];AIF胞核阳性的细胞具有凋亡细胞的特征,阳性细胞的数量与脑梗死的面积呈线性关系,且不受caspase的抑制剂BAF的影响,说明AIF介导的caspase非依赖性凋亡途径在新生鼠脑缺血再灌注损伤的发病过程中发挥作用。
L-NAT是P物质(substance P,SP)的特异性受体 neurokinin 1 receptor(NK-1)的拮抗剂[12],是经过美国FDA批准用于治疗恶心、呕吐、休克及神经退行性病变的药物。已有研究表明,L-NAT可通过抑制CytC的释放而抑制线粒体途径的细胞凋亡[13]。本研究用H2O2诱导PHNs制备海马神经元细胞凋亡模型,研究发现H2O2诱导后,细胞失去原有形态,胞体肿胀,突起减少或消失,细胞培养上清液中LDH的量增多,细胞活性及存活率降低,而L-NAT组神经元的胞体较完整,大部分细胞存在少量的突起,L-NAT可降低H2O2诱导后神经细胞中LDH的分泌,提高细胞的存活率,说明 L-NAT对H2O2诱导后PHNs细胞凋亡具有保护作用。本研究还发现,L-NAT还可减少活化型caspase-3的表达和caspase-3的活性、抑制CytC和AIF等线粒体促凋亡因子从线粒体向胞质的释放、并恢复了部分细胞中Rh123阳性产物在胞质中的颗粒性分布,说明L-NAT可抑制线粒体膜势能的丧失、通过抑制caspase依赖性和非依赖性的细胞凋亡途径,对缺血再灌注损伤的神经细胞产生保护作用。
[1]Ueda H,Matsunaga H,Uchida H,et al.Prothymosin alpha as robustnessmolecule against ischemic stress to brain and retina[J].Ann N Y Acad Sci,2010,1194:20 - 26.
[2]Mehta SL,Manhas N,Raghubir R.Molecular targets in cerebral ischemia for developing novel therapeutics[J].Brain Res Rev,2007,54:34 -66.
[3]Zhang WH,Wang H,Wang X,et al.Nortriptyline protects mitochondria and reduces cerebral ischemia/hypoxia injury[J].Stroke,2008,39:455 -462.
[4]Vosler PS,Graham SH,Wechsler LR,et al.Mitochondrial targets for stroke:focusing basic science research toward development of clinically translatable therapeutics[J].Stroke,2009,40:3149 -3155.
[5]Cheng J,Wang F,Yu DF,et al.The cytotoxicmechanism of malondialdehyde and protective effect of carnosine via protein cross-linking/mitochondrial dysfunction/reactive oxygen species/MAPK pathway in neurons[J].Eur JPharmacol,2011,650:184 -194.
[6]Galluzzi L,Blomgren K,Kroemer G.Mitochondrial membrane permeabilization in neuronal injury[J].Nat Rev Neurosci,2009,10:481 -494.
[7]Shoshan-Barmatz V,Keinan N,Abu-Hamad S,etal.Apoptosis is regulated by the VDAC1 N-terminal region and by VDAC oligomerization:release of cytochrome c,AIF and Smac/Diablo[J].Biochim Biophys Acta,2010,1797:1281-1291.
[8]Wang X,Figueroa BE,Stavrovskaya IG,etal.Methazolamide and melatonin inhibit mitochondrial cytochrome C release and are neuroprotective in experimentalmodels of ischemic injury[J].Stroke,2009,40:1877 -1885.
[9]Zem lyak I,Brooke SM,Singh MH,et al.Effects of overexpression of antioxidants on the release of cytochrome c and apoptosis-inducing factor in the model of ischemia[J].Neurosci Lett,2009,453:182 -185.
[10] M illan A,Huerta S.Apoptosis-inducing factor and colon cancer[J].JSurg Res,2009,151:163 -170.
[11]Zhu C,Qiu L,Wang X,et al.Involvement of apoptosis-inducing factor in neuronal death after hypoxia-ischemia in the neonatal rat brain[J].JNeurochem,2003,86:306 -317.
[12]Turner RJ,Helps SC,Thornton E,et al.A substance P antagonist improves outcomewhen administered 4 h after onset of ischaemic stroke[J].Brain Res,2011,1393:84 -90.
[13]Wang X,Zhu S,Pei Z,et al.Inhibitors of cytochrome c release with therapeutic potential for Huntington's disease[J].JNeurosci,2008,28:9473-9485.
猜你喜欢
建材发展导向(2021年11期)2021-07-28
当代水产(2020年10期)2020-03-17
野生动物学报(2020年1期)2020-02-21
当代水产(2019年8期)2019-10-12
西南农业学报(2017年6期)2017-08-08
中国高原医学与生物学杂志(2017年4期)2017-03-08
天津医药(2016年9期)2016-10-20
中国组织化学与细胞化学杂志(2016年3期)2016-02-27
中国老年学杂志(2015年16期)2015-03-05
癌变·畸变·突变(2015年4期)2015-02-27
