
荧光显微图像亚细胞斑点检测方法研究进展
2012-12-31 13:17谭映军李莹辉郑筱祥
中国生物医学工程学报 2012年6期
吴 坚 赵 挺 谭映军 李莹辉 郑筱祥,*
1(浙江大学生物医学工程与仪器科学学院,生物医学工程教育部重点实验室,浙江省心脑血管
检测技术与药效评价重点实验室,杭州 310027)2(浙江大学求是高等研究院,杭州 310027)3(中国航天员科研训练中心,航天医学基础与应用国家重点实验室,北京 100094)
引言
随着显微镜和信息处理技术的发展,越来越多的科技工作者使用高性能荧光显微镜(LSCM、双光子、TRIFM等)观察亚细胞结构成分(蛋白质囊泡、染色体、微丝等)及其动力学特性(囊泡迁移、融合、有丝分裂等),得到海量的图像数据[1-6]。人工处理这些数据不仅工作十分繁琐,同时受到成像质量不高、观察对象特征复杂、人工诊断主观性较强、观察者视觉疲劳等因素的影响,分析结果往往假阳性率和假阴性率高,所以借助图像处理与分析技术提供客观的定量数据,帮助量化并验证所观察到的生命过程,就显得尤为必要[7-11]。特别是在蛋白质组学、功能性基因组学和药物筛选等生物学研究领域,自动分析处理高通量数据具有重要意义[12-13]。
生物显微图像处理首先要解决的是自动准确检测观察对象,例如解释复杂的细胞显型常常需要知道特定细胞器在细胞中的位置、数量、聚集度等参数[14];利用共聚焦显微镜观察得到的细胞图像进行计算机三维重构,依赖准确检测亚细胞结构成分[15];活细胞序列成像中,追踪特定分子探针标记的亚细胞目标,需要在每帧图像中确定追踪对象的准确位置[16-18]。被特定荧光探针标记的亚细胞观察对象,在细胞图像中常以模糊亮斑的形式出现,其亮度高于背景,面积相对较小又较为致密,且没有明显的边界,称之为亚细胞斑点。目前常用的生物图像处理软件(如ImageJ等)更多的是采用阈值法来检测斑点。虽然简便,但没有经过滤波处理,过度依赖图像质量和阈值的选取,效果往往很不理想。
文中就生物荧光显微图像中常见的亚细胞斑点问题,在分析荧光图像中所含的噪声类型和图像形成过程的基础上,对其检测方法进行综述,按照设计思想对方法加以归纳、总结、分类,最后总结讨论了各检测方法的优缺点和该研究领域仍然存在的难点。
1 荧光显微图像中的噪声
由于成像过程的局限,得到的图像往往信噪比较低,而且图像中斑点数量较多,聚集度高,相互粘连,且常常淹没在周围复杂的背景中,如图1(a)所示原代脂肪细胞中的Glut4囊泡。在活细胞荧光显微成像时,为了防止荧光淬灭和荧光染料化学损伤,光照强度常被设置得很低,所以得到的图像信噪比更低,如图1(b)所示活细胞工作站拍摄的C2C12骨骼肌细胞中的Glut4囊泡。生物荧光显微图像中强噪声干扰的存在使得自动斑点检测极具挑战性[11,19-21]。

图1 细胞图像中的Glut4囊泡斑点。(a)原代脂肪细胞;(b)C2C12骨骼肌细胞Fig.1 The Glut4 vesicle spots in cell images.(a)A primary adipocyte;(b)A C2C12muscle cell
显微成像仪器设计制造,一直致力于改进由于相位失真和光学相差而引入的干扰。然而,在实际应用中干扰是无处不在的,没有哪一个光学系统可以完全避免。成像系统中显微镜的光源、物镜、分光镜、光敏检测器等关键部件以及显微镜的类型,都需要很好地调整和选择才能获得高信噪比的锐利图像。既使所有的部件都已调试得很好,各部件的物理局限还是会给图像带来固有噪声,主要有:因光子产生的随机性,给光敏检测器带来的Poisson型光子散射噪声;由于诱发电子热搅动引起剧烈震荡所形成的暗流,温度越高,暗流也越剧烈,同样服从Poisson分布;电子设备在数据读取时会产生读出噪声,幅值与读取速率逆相关,功率谱密度随着1/f递减,表现为加性Gaussian噪声;幅值量化过程中的量化噪声是A/D转换设备固有的[22],与信号无关,实际使用中可将其控制在其他噪声均方根一半以内[11]。
此外,荧光染料的光学性质与成像密切相关,同样也会成为影响图像质量的重要因素,如有机或无机分子的自发荧光是图像中常见的背景干扰。同时,光在激发和发射时由于粒子性会发生散射,在不同介质间传输会发生折射,所以随着样本观察的深入,激发光被样本材料吸收导致样本发射荧光能力减弱。
2 荧光显微成像中的图像形成
目前,广泛使用的荧光显微镜都是落射成像系统,其激发光和发射光通过同一物镜,成像过程是非相干的。当激发光照射到样本上时,样本中的每个荧光团分子被激发作为第二光源(点光源),通过物镜成像并被光敏检测器检测到,其对图像亮度分布的贡献是独立的。显微成像系统的点扩散函数(point spread function,PSF)是点光源通过物镜成的像,即物镜系统的冲击响应。理论上PSF能够表示为标量Debye衍射积分,也可以表示为出瞳函数(pupil function)的二维Fourier变换。例如波长为λ的窄带非相干点光源通过直径为a的圆形孔径物镜所形成的PSF是圆形对称的,可以表示为
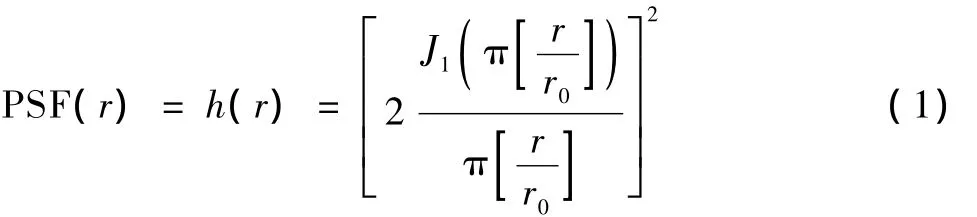
式中,J1是第一类一阶Bessel函数,常数r0=λdi/a是尺度因子,di为像距,径向距离r代表像中的每个点与像平面原点的距离。
实际上PSF可以根据图像数据的维数用二维或三维Gaussian函数非常准确地近似[23-24],如图2所示:其中图(a)尺寸为31像素×31像素,中心亮度为255;图(b)尺寸为91像素×91像素,点光源坐标为(46,46),亮度为255;图(c)为点光源与PSF卷积所成的像(斑点),尺寸同图(b)。
因此,荧光显微成像系统可以建模为线性空间移不变系统,图像的形成就是每一个点光源成像的线性集合,那么图像亮度分布可以表示为式中,M是物镜的放大倍数,χ函数与荧光团有关,描述样本在激发光下发射波长为λ的荧光的能力。
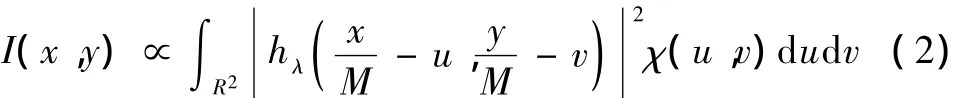
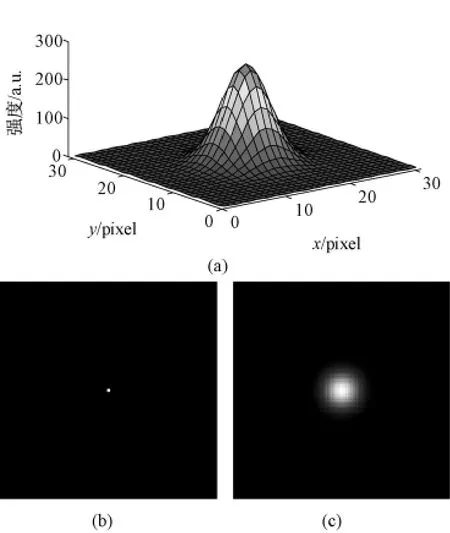
图2 二维Gaussian函数模拟的非相干点扩散函数。(a)点扩散函数三维示意图;(b)点光源;(c)点光源的像Fig.2 The incoherent PSF simulated with 2D Gaussian distribution.(a)3D PSF schematic diagram;(b)A point source;(c)An image of point source(b)
3 斑点检测方法
除了简单的阈值法外,常见的斑点检测方法都是通过分析荧光显微图像的形成过程构造合适的函数模拟PSF,考虑图像的噪声类型选用合适的降噪技术,针对斑点的形状和大小选择合适的参数。检测流程大体上包括降噪滤波、信号增强、信号阈值化等3个步骤。降噪滤波初步去除图像包含的噪声(灰度图像),信号增强进一步增强目标信号抑制背景干扰和噪声(灰度图像),信号阈值化通过设置硬或软阈值提取检测目标(二值图像)。在实际操作过程中,不同的检测方法在每一步执行上又有所区别,一些步骤是可以选择或合并的。例如有的方法自身带有降噪的效果,不需要添加额外的降噪步骤,但是所有的检测方法都离不开信号增强,因为这是各检测方法最显著的区别。整体检测流程如图3所示。
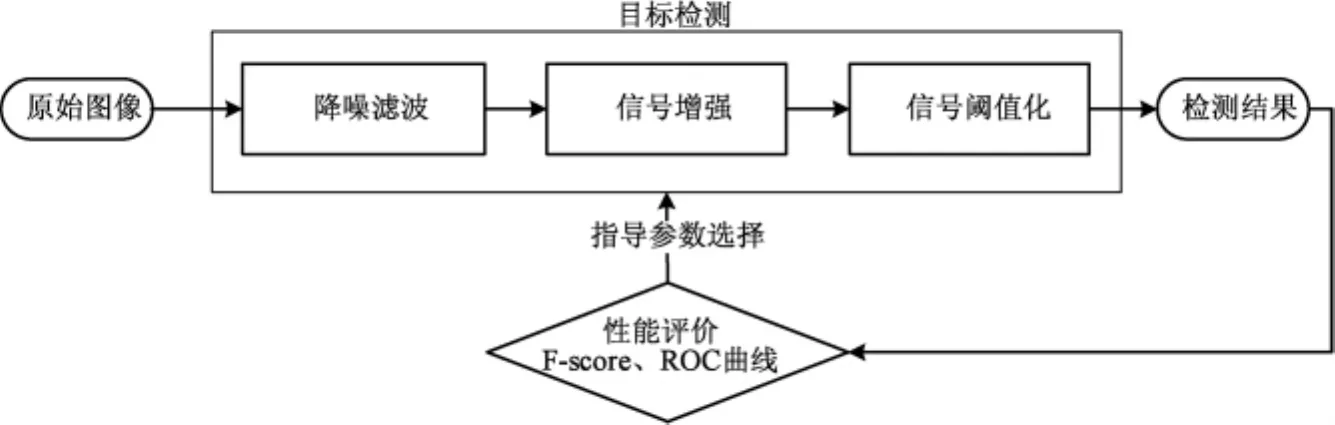
图3 斑点检测流程Fig.3 The spot detection framework
3.1 降噪滤波
去除原图像中的噪声能有效改善信噪比,提高图像质量和目标的可检测性。荧光显微图像中噪声的主要来源是Poisson型光子散射噪声和加性Gaussian型读出噪声。前者是光子发射随机性引起的,虽然可以通过提高光照强度或增加曝光时间被极大地降低,但会引起荧光漂白现象;后者是检测器件的电子特性引起的;两者相互独立。大多数情况下,降噪处理使用计算复杂度较低的高斯平滑(Gaussian smoothing)[25]或匹配滤波(match filtering)[26]。一些非线性滤波技术,如多尺度小波降噪[27]、Patch-based降噪[28]、方差稳定变换(variance stabilizing transform,VST)[29]、形态学滤波[30]等也在一些文献报道中出现。特别是文献[29]将Poisson型或Poisson-Gaussian混合型噪声转变为近似Gaussian型噪声来处理,能有效去除荧光显微图像中的光子散射噪声和读出噪声。
3.2 信号增强
信号增强是在降噪后的图像中,进一步增强目标信号同时抑制不相关的背景。在这个步骤中并没有检测出目标或图像特征,因为提取不出可以量化的数据,如目标的位置、个数、面积等信息,仍需要依靠后续处理连接属于同一目标的像素。目前采用的方法大多数是无监督型的,通过显式或隐式假设目标模型,手动或半自动调整参数达到应用于特定情况下的最佳效果。文献[31]将各种方法按照数据驱动型和模型驱动型分为从底至顶(from bottom to top)和从顶至底(from top to bottom)两类。本文选择其中8种常用方法,按照它们的设计思想概括为基于匹配滤波的、基于数学形态学的和基于小波多尺度等3类加以介绍。表1列举了各方法的特点。

表1 斑点检测方法Tab.1 The spot detection methods
3.2.1 基于匹配滤波的方法
匹配滤波方法主要是通过模拟PSF,将滤波后的图像与类点扩散函数核进行卷积运算,然后在斑点存在区域(其图像亮度分布与核匹配)内产生高强度响应,而在图像的其他区域内只有低强度响应,以达到增强目标和抑制不相关背景的目的。该类方法的参数需要针对斑点的尺寸合理选择。最为常见的是采用Gaussian核,以下是对其有效改进后的3种方法。
(1)斑点增强滤波器
文献[32]提出的斑点增强滤波器(spot enhancing filter,SEF),使 用 Laplacian of Gaussian(LoG)核取代Gaussian核近似白化匹配滤波器来增强目标抑制噪声。原图像I与LoG核(2-i2-j2)GσL卷积,得到目标增强图像 C,其中 GσL是Gaussian核,参数 σL是 L oG核宽,与斑点的尺寸大小有关。最后,通过信号阈值化操作提取斑点目标。SEF将降噪滤波和信号增强两步结合在一起实现,计算简便,但提取目标过度依赖阈值选取。
(2)亚像素定位检测
文献[16]提出的亚像素定位(sub-pixel location,SPL)检测是序列图像中目标追踪的关键步骤。SPL首先通过相邻w帧图片的时间平均有效降低背景噪声干扰;然后逐点在3像素×3像素区域内初步找出图像的局部最大值,并规定该最大值满足背景标准差的α倍时有效;最后在每个有效的局部最大值位置对原图像I进行混合Gaussian模型迭代拟合逐个提取斑点。在某一局部最大值处,当拟合完第k+1个核后与原图像的差异在统计意义上明显小于拟合k(≥1)个核后的差异时,拟合继续,否则拟合结束,具体参见文献[24]。SPL主要是针对追踪问题设计的,它只能提供目标的位置信息(有效局部最大值),可以用来统计图像中斑点的个数,不能进行斑点像素级分析,得益于Gaussian迭代拟合,可检测粘连目标。
(3)特征点检测
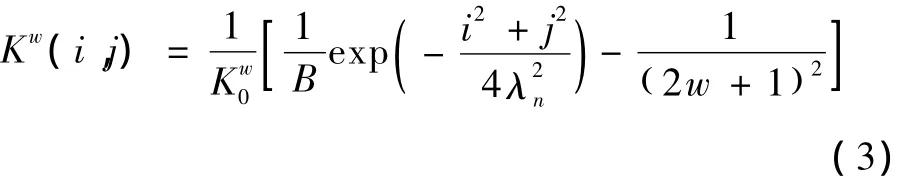
文献[17]提出的特征点检测(Feature Point Detection,FPD)也来源于目标追踪问题,最初设计用于检测胶质颗粒[33]。FPD首先选择合适参数的Gaussian卷积核对原图像I进行复原,其中Kw0和B是归一化常数,λn和w分别定义Gaussian核宽和可调核窗宽度,w取值应大于最大斑点半径小于斑点间最小距离;然后通过灰度形态学膨胀操作提取图像的局部最大值,用mean-shift方法结合周围像素值优化局部最大值位置;最后分别计算每个局部最大值对应点集(中心为局部最大值,半径为w)亮度的一阶矩和二阶矩,以此来判断该点集是否对应检测目标。FPD和SPL一样,也只能得到斑点的位置信息(优化的局部最大值)。
3.2.2 基于数学形态学的方法
数学形态学处理依靠膨胀、腐蚀、开和闭等4种基本操作的灵活组合,可以实现包括图像滤波、分割、测量、以及纹理分析和合成等功能。该类检测操作针对斑点的形态构建合适的结构元素,通过相应的形态学运算,将其从局部不相关背景中提取出来。结构元素的选取与斑点的形状、大小和方向等先验知识有关。
(1)灰度开运算顶帽变换
文献[34]提出的灰度开运算顶帽(grayscale opening top-hat,GOTH)变换,利用开操作去除相关的斑点图像目标,再通过开图像与原图像之间的算术差分运算提取斑点,具体做法如下:先用核宽为σ的Gaussian核对原图像I进行滤波,得到滤波图像J;再用半径为r的平圆盘结构元素A对J进行开操作得到JA,其中结构元素的半径与最大斑点的尺寸有关;最后滤波图像J与JA进行差分运算,即J-JA得到目标增强图像C,最后通过信号阈值化提取斑点目标。该方法与试图直接抑制不相关背景提取目标的思路相反,其在低信噪比图像中检测效果不好,过度依赖阈值选取。
(2)H-Dome变换
文献[18]提出的H-Dome变换,也是用于追踪过程的目标检测,实质上是提取亮度局部最大值的形态学H-maxima操作[11],只是做了s次幂的扩展,并进行检测结果的判别。该方法可以分为滤波、形态学重构、结果判别3个步骤,具体操作如下:原图像I通过核宽为σL的LoG核滤波后得到滤波图像J;将J-h作为标记对J进行形态学重构得到重构图像B,其中h>0是常数,与斑点和相应背景的最小亮度差有关。H-Dome图像为
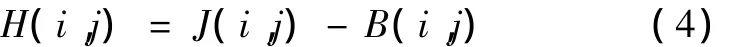
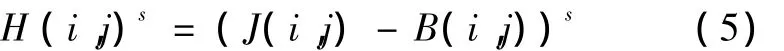
对H中的每点像素值提升s次幂可以有效增强目标抑制背景,即再通过Important Resampling算法从H(i,j)s中重新采样N个像素点,用mean-shift算法聚类成M个点集对应检测目标,最后计算每个点集对应协方差矩阵行列式的值与给定阈值σM比较判断该点集是否有效,并得到目标的位置信息(点集mean-shift聚类中心)。
(3)基于旋转操作的顶帽变换
文献[30]介绍的方法除了可用于图像滤波外,也可以用于检测斑点。该方法基于旋转操作的顶帽(rotational morphological processing based top-hat,RMPTH)变换,采用直线片段结构元素L(1×N),N为长度。考虑到在离散空间中旋转结构元素很难在任意角度产生直线结构,所以采用将原图像I顺时针旋转πi/M[rad](i=0,1,…,M-1)后与固定位置的结构元素L进行开操作得到Oi,再将M个Oi相应逆时针旋转πi/M[rad]得到O'i,最终RMPTH变换结果
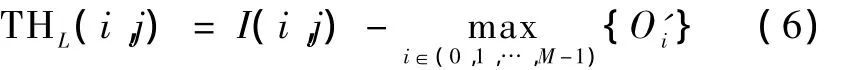
对应目标增强图像C,最后通过信号阈值化提取斑点目标。得益于单像素宽度的结构元素,该方法能够区分粘连的斑点。
3.2.3 基于小波多尺度的方法
多尺度小波分析将空域局部性和频域局部性相结合,使其可以对信号进行稀疏表达。这种稀疏性在图像数据压缩、噪声滤波和图像特征检测中得到广泛应用。该类方法主要是利用特定小波变换将图像分解为多个尺度分量,然后分别对相应尺度分量进行软或硬阈值操作,再将尺度分量部分相加或相乘,以达到噪声过滤和目标检测的目的。
(1)小波多尺度乘积
文献[27]采用基于 B 3样条([1,4,6,4,1]/16)的à trous小波对原图像I进行K层多尺度分解,得到细节图像(detail image)Wk(i=1,…,K)和近似图像(approximation image)IK,有
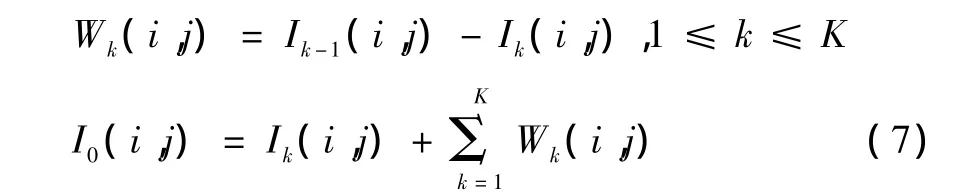
式中,Ik是Ik-1与B3样条逐行逐列卷积的结果。每层卷积过后B3样条相邻元素间要插入2k-1-1个0以适应下一个尺度的计算。Wk(k=1,…,K)通过计算其中值绝对偏差(median absolute deviation,MAD)进行自适应阈值化得到去噪的~Wk,再将K个尺度的~Wk相乘(wavelet multiscale product,WMP)得到检测目标增强图像C最后通过灰度阈值化提取目标。
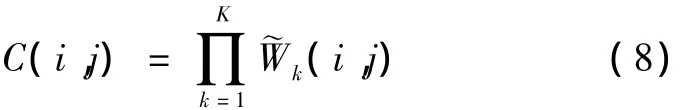
(2)多尺度方差稳定变换
文献[35]在文献[29]基础上针对荧光显微图像包含Poisson型噪声,提出多尺度方差稳定变换(multiscale variance stabilizing transform,MSVST),对WMP进行了改进,主要的改进有3点:1)将原图像I通过VST处理;2)通过控制错误发现率(false discovery rate,FDR)进行Wk阈值操作;3)用混合最速下降(hybird steepest descent,HSD)迭代重构图像。该文还针对小波基在二维尺度上方向性不足引入脊波(ridgelet)和曲波(curvelet),改进了MSVST。
除以上介绍的方法外,文献[36]中介绍的KDE、LC、LEF、SE等方法在特定的场合也得到了应用。文献[37-38]中提出的Ada-Boost和Fisher判别分析这类机器学习方法,用于目标检测也取得了不错的效果,但需要在注释过的训练样本中学习得到目标的特征,再利用提取的特征检测目标。由于生物图像的复杂性,构建合适的训练样本集是困难的。
3.3 信号阈值化
为了从增强过的图像中得到斑点的像素级信息,需要引入阈值化操作。先将目标增强图像C经过灰度阈值ld得到二值图像CB,利用CB作为掩模在原始图像中标记目标并做亮度分析,可得到斑点的整体亮度、平均亮度、面积、周长、圆度、长短轴长、质心等定量数据。由于灰度阈值对目标的提取影响较大,所以使用尺寸阈值vd=(vmax,vmin)加以约束。只有当图像中尺寸大于最小阈值vmin且小于最大阈值vmax的点集才被认为是检测到的斑点。由于图像噪声并没有被完全去除,阈值化得到的掩模图像CB点集区域的非零像素点并不完全相连,点集中包含有错误的零像素点,这个问题可以使用形态学闭运算解决[11,34]。
4 检测方法的性能评价标准
评价各类检测方法的性能首先需要知道图像中斑点的位置、个数、面积大小等信息的真实参考(ground truth)。这些只能靠生物学家人工分析显微图像获得,也就是金标准(gold standard)。当图像的真实参考已知时,对检测结果的真阳性(true positive,表示检测结果符合生物学意义)、假阳性(false positive,表示检测结果不符合生物学意义)、假阴性(false negative,表示未被检测出的符合生物学意义)、真阴性(true negative,表示未被检测出的不符合生物学意义)可以很容易进行判别。此时,有很多性能评价标准存在[39-40]。这里介绍两类常见的评价标准:F-score和ROC曲线。
(1)F-score
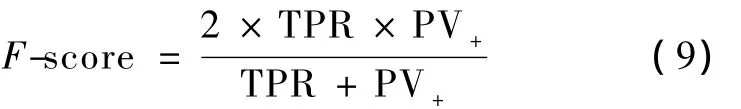
结合阳性预测值PV+和真阳性率TPR定义的度量标准F-score为式中,PV+表示TP占检测结果(TP+FP)的比例,TPR表示TP占真实参考(TP+FN)的比例,F-score可更全面地评价目标检测性能,值越大性能越好。
(2)ROC曲线
接收者工作特性曲线(receiveroperating characteristic curve,ROC curve)是以假阳性率FPR(FP占FP+TN的比例)为横坐标,真阳性率TPR为纵坐标,横轴和纵轴的长度相等,形成正方形,在图中将ROC工作点标出,用直线连接各相邻两点构建的非光滑曲线。理论上,当检测结果完全没有意义时,有TPR=FPR,是一条从原点到右上角的对角线,称为机会线(chance line)。ROC曲线一般位于机会线的上方,离机会线越远,说明检测准确度越高。最好的检测方法表现为ROC曲线从原点垂直上升至左上角,然后水平到达右上角。ROC曲线下的面积AROC也可以反映检测方法的性能,取值范围为(0.5,1),完全没有价值的检测AROC=0.5,完全理想的检测AROC=1。一般认为AROC在0.5到0.7之间表示检测准确性较低,0.7到0.9时准确性中等,大于0.9准确性较高[41]。
5 总结展望
本文所介绍的亚细胞斑点检测方法都是针对特定应用场合设计的,具体方法需要根据不同的应用背景进行合理的选择。其中,由于计算简便被广泛使用的SEF和GOTH,需要通过信号阈值化操作得到斑点的二值掩模图像标记原图像,以此进行后续的像素级信息统计,所以对噪声和阈值的选取比较敏感;SPL、FPD和H-Dome是为目标追踪问题而设计的,只考虑检测斑点的位置信息,不需要通过信号阈值化操作提取检测目标,其中FPD和HDome因为加入结果判别准则,有效地提高了检测准确度;WMP和MSVST都隐式含有滤波处理,不需要额外的滤波操作,提取目标虽然不需要尺寸阈值化,但都包含灰度阈值化操作,其中MSVST对于处理Poisson-Gaussian混合噪声效果显著;RMPTH和SPL得益于直线片段结构元素和Gaussian迭代拟合,能够检测出粘连的斑点;H-Dome和MSVST算法复杂度较高,可用于信噪比极低的图像数据,文献[42]的结论表明在测试数据中,当信噪比小于2时,H-Dome和MSVST检测仍然有效且效果不相上下。
虽然现代光学显微成像技术已经取得了很大的进步,但和亚细胞结构成分的尺寸(约几个nm)相比,目前最先进显微镜的分辨率(约100 nm)仍然很低[43-44]。除了成像过程引入的固有噪声,染料选取、染色方式以及显微镜操作不当,都会造成采集到的图像信噪比较低、均一度差。在这种情况下,即使专业的生物学家将目标从噪声和不相关背景中区分开也是相当困难的。在序列成像过程中,上述问题更为严重。除了问题本身的难度,现有方法的瓶颈在于只适用某种成像条件或某种细胞器类型,研究者无法全面了解问题,所提出方法的通用性受到了很大的限制。导致目前各种算法实用性不强的共性难点有:(1)现有内置或外在的滤波技术很难完全去除复杂的噪声干扰,对后续操作影响较大;(2)每种算法针对不同图像都有多个与检测目标密切相关的参数需要设置,不能做到自适应调节;(3)缺乏可靠的检测结果评价标准,方法间不能客观地进行比较。这些都给用户选择合适的方法带来了巨大困难。
总之,要解决目前荧光显微图像目标检测中存在的问题,与成像技术的发展和数据产生的速度相适应,需要在每个环节上有所创新和突破。首先,建立全面可靠的基准测试库。目前科研人员已经开始着手收集细胞和各类生物样本的基准测试图样,对检测方法进行比较和检验[45-47]。其次,采用机器学习的方法针对特定目标样本图像训练得到自适应的算法参数。最后,提出更能客观反映算法性能的评价标准,并以此为依据研究鲁棒性和通用性更强的新方法。
[1]Lippincott-Schwartz J,Patterson GH.Development and use of fluorescent protein markers in living cells[J].Science,2003,300(5616):87-91.
[2]Miyawaki A,Sawano A,Kogure T.Lighting up cells:Labelling proteins with fluorophores[J].Nature Cell Biology,2003,5(Suppl):1-7.
[3]Stephens DJ,Allan VJ.Light microscopy techniques for live cell imaging[J].Science,2003,300(5616):82-86.
[4]Tsien RY.Imagining imaging’s future[J].Nature Cell Biology,2003,5(Suppl):16-21.
[5]Pepperkok R,Ellenberg J.High throughputfluorescence microscopy for systems biology[J].Nature Reviews Molecular Cell Biology,2006,7(9):690-696.
[6]Vonesch C,Aguet F,Vonesch JL,et al.The colored revolution of bioimaging[J].IEEE Signal Processing Magazine,2006,23(3):20-31.
[7]Eils R,Athale C.Computational imaging in cell biology[J].The Journal of Cell Biology,2003,161(3):477-481.
[8]Zhou X,Wong STC.Informatics challenges of high throughput microscopy[J].IEEE Signal Processing Magazine,2006,23(3):63-72.
[9]Shorte SL,Frischknecht F.Imaging Cellular and Molecular Biological Functions[M].Berlin:Springer-Verlag,2007:45-70.
[10]Ahmed WM,Leavesley SJ,Rajwa B,et al.State of the art in information extraction and quantitative analysis for multimodality biomolecular imaging[J].Proceedings of the IEEE,2008,96(3):512-531.
[11]WuQ,MerchantFA,Castleman KR.MicroscopeImage Processing[M].Burlington:Elsevier,2008:247-297.
[12]Drubin DA,Garakani AM,Silver PA.Motion as a phenotype:The use of live-cell imaging and machine visual screening to characterize transcription dependent chromosome dynamics[J/OL].BMC CellBiology,2006,7(19).http://www.biomedcentral.com/1471-2121/7/19,2006-04-24/2012-07-18.
[13]Neumann B,Held M,Liebel U,et al.High-throughput RNAi screening by time-lapse imaging of live human cells[J].Nature Methods,2006,3(5):385-390.
[14]Sacher R,Stergiou L,Pelkmans L.Lessons from genetics:interpreting complex phenotypes in RNAi screens[J].Current Opinion Cell Biology,2008,20(4):483-489.
[15]Khodade P,Malhotra S,Kumar N,etal.Cytoview:development of a cell modelling framework[J].Journal of Bioscience,2007,32(5):965-977.
[16]Jaqaman K,Loerke D,Mettlen M,et al.Robust single-particle tracking in live-cell time-lapse sequences[J].Nature Methods,2008,5(8):695-702.
[17]Sbalzarini IF,Koumoutsakos P.Feature point tracking and trajectory analysis for video imaging in cell biology[J].Journal of Structure Biology,2005,151(2):182-195.
[18]Smal I,Meijering E,Draegestein K,et al.Multiple object tracking in molecular bioimaging by rao-blackwellized marginal particle filtering[J].Medical Image Analysis,2008,12(6):764-777.
[19]Dorn JF,Danuser G,Ge Y.Computational processing and analysis of dynamic fluorescence image data[J].Methods in Cell Biology,2008,25:497-538.
[20]Gerlich D,Ellenberg J.4D imaging to assay complex dynamics in live specimens[J].Nature Cell Biology,2003,5(Suppl):14-19.
[21]MeijeringE,SmalI,DanuserG.Tracking in molecular bioimaging[J].IEEE Signal Processing Magazine,2006,23(3):46-53.
[22]Stevens JK,Mills LR,Trogadis JE.Three Dimensional Confocal Microscopy:Volume Investigation of Biological Specimens[M].London:Academic Press,1994:47-93.
[23]Zhang B,Zerubia J,Olivo-Marin J-C.Gaussian approximations of fluorescence microscope point spread function models[J].Applied Optics,2007,46(10):1819-1829.
[24]Thomann D,Rines DR,Sorger PK,et al.Automatic fluorescent tag detection in 3D with super-resolution:Application to the analysis of chromosome movement[J].Journal of Microscopy,2002,208(1):49-64.
[25]RomenyTHBM.Front-End Vision and Multi-Scale Image Analysis[M].Berlin:Springer,2003:277-284.
[26]Turin GL.An introduction to matched filters [J].IRE Transactions on Information Theory,1960,6(3):311-329.
[27]Olivo-Marin J-C.Extraction of spots in biological images using multiscale products[J].Pattern Recognition,2002,35(9):1989-1996.
[28]Boulanger J,Kervrann C,Bouthemy P.Patch-based nonlocal functional for denoising fluorescence microscopy image sequences[J].IEEE Transactions on Medical Imaging,2010,29(2):442-454.
[29]Zhang B,Fadili J,Starck JL.Multiscale variance-stabilizing transform for mixed-Poisson-Gaussian processes and its applications in bioimaging [C].Proceedings of IEEE International Conference on Image Processing.San Antonio:IEEE,2007,6(Ⅵ):233-236.
[30]KimoriY,Baba N,Morone N.Extended morphological processing:a practical method for automatic spot detection of biological markers from microscopic images[J/OL].BMC Bioinformatics,2010,11(373).http://www.biomedcentral.com/1471-2105/11/373,2010-07-08/2012-07-18.
[31]RohrK,GodinezWJ,HarderN,etal.Tracking and quantitative analysis of dynamic movements of cells and particles[J/OL].Cold Spring Harb Protocols,2010,6:1-16.http://cshprotocols.cshlp.org/content/2010/6/pdb.top80.full,2010-06-01/2012-07-18.
[32]Sage D,Neumann FR,Hediger F,et al.Automatic tracking of individual fluorescence particles:Application to the study of chromosome dynamics [J].IEEE Transactionson Image Processing,2005,14(9):1372-1383.
[33]Crocker JC,Grier DG.Methods of digital video microscopy for colloidal studies[J].Journal of colloid and interface science,1996,179:298-310.
[34]Soille P.Morphological Image Analysis:Principles and Applications[M].Berlin:Springer-Verlag,2003:121-127.
[35]Zhang B,Fadili J,Starck J.Wavelets,ridgelets,and curvelets for Poisson noise removal[J].IEEE Transactions on Image Processing,2008,17(7):93-108.
[36]Ruusuvuori P,Äijö T,Chowdhury S,et al.Evaluation of methods for detection of fluorescence labeled subcellular objects in microscope images[J/OL].BMC Bioin for matics,2010,11(248).http://www.biomedcentral.com/1471-2105/11/248,2010-05-13/2012-07-18.
[37]Jiang S,Zhou X,Kirchhausen T,et al.Detection of molecular particles in live cells via machine learning[J].Cytometry A,2007,71(8):563-575.
[38]Mclachlan GJ.Discriminant Analysis and Statistical Pattern Recognition[M].New York:Wiley,2004:129-336.
[39]Fawcett T.An introduction to ROC analysis[J].Pattern Recognition Letters,2006,27:861-874.
[40]Popovic A,De La Fuente M,Engel hardt M,et al.Statistical validation metric for accuracy assessment in medical image segmentation[J].International Journal of Computer Assisted Radiology and Surgery,2007,2:169-181.
[41]Swets JA.Measuring the accuracy of diagnostic systems[J].Science,1988,240:1285-1293.
[42]Smal I,Loog M,Niessen W,et al.Quantitative comparison of spot detection methods in fluorescence microscopy[J].IEEE Transactions on Medical Imaging,2010,29(2):282-301.
[43]Hell SW,Dyba M,Jakobs S.Concepts for nanoscale resolution in fluorescence microscopy[J].Current Opinion in Neurobiology,2004,14(5):599-609.
[44]Garini Y,Vermolen BJ,Young IT.From micro to nano:Recent advances in high-resolution microscopy[J].Current Opinion in Neurobiology,2005,16(1):3-12.
[45]Ruusuvuori P,Lehmussola A,Selinummi J,et al.Benchmark set of synthetic images for validating cellimage analysis algorithms[C]//Proceedings of the 16th European Signal Processing Conference.Lausanne: EUSIPCO,2008:1569105508.
[46]Broad bioimage benchmark collection[DB/OL].http://www.broad.mit.edu/bbbc,2012-05-21/2012-07-18.
[47]Gelasca ED,Byun J,Obara B,et al.Evaluation and benchmark for biological image segmentation[C]//Proceedings of IEEE International Conference on Image Processing.San Diego:IEEE,2008:1816-1819.
猜你喜欢
新作文·小学低年级版(2022年3期)2022-08-30
智慧少年·故事叮当(2021年5期)2021-08-23
临床骨科杂志(2020年1期)2020-12-12
制造技术与机床(2019年9期)2019-09-10
今日农业(2019年15期)2019-01-03
电子制作(2018年16期)2018-09-26
火控雷达技术(2016年3期)2016-02-06
探测与控制学报(2015年4期)2015-12-15
海军航空大学学报(2015年1期)2015-11-11
空间控制技术与应用(2015年3期)2015-06-05
