
新型手性四氮镍(Ⅱ)配合物的合成、表征及催化苯乙酮不对称氢转移研究
2012-12-22 07:34谭华杰洪益玲
杭州师范大学学报(自然科学版) 2012年1期
张 添,谭华杰,洪益玲,沈 良
(杭州师范大学材料化学与化工学院,浙江杭州 310036)
新型手性四氮镍(Ⅱ)配合物的合成、表征及催化苯乙酮不对称氢转移研究
张 添,谭华杰,洪益玲,沈 良
(杭州师范大学材料化学与化工学院,浙江杭州 310036)
合成了两个手性四氮配体,(S,S,S,S)-N,N′-二[2-(对甲苯磺酰胺基)-1,2-二苯基乙基]乙二胺(A)和(S,S,S,S)-N,N′-二[2-(对甲苯磺酰胺基)-1,2-二苯基乙基]丙二胺(B),及其手性镍(Ⅱ)配合物1和2,采用元素分析、1H NMR、MS和IR对它们进行了表征.研究了手性配合物催化苯乙酮的不对称氢转移反应,考察了碱量和反应时间对催化活性的影响.
手性四氮配体;镍配合物;苯乙酮;不对称氢转移
近年来,随着化工、医药等领域对光学纯手性醇中间体需求的不断增加,不对称氢转移(asymmetric transfer hydrogenation,ATH)作为具有独特优势——催化效率高、操作简便、宽的底物适应性——的合成方法[1-5],受到广泛关注.ATH所要求的核心条件是高效、低耗、经济的手性配合物作为催化剂,在适当的温度、碱量、底物/催化剂(S/C)条件下进行.
手性配体对不对称氢转移获得高转化率和高ee值起着非常重要的作用,在众多配体中,四氮配体吸引了我们的目光,基于其以下优点:易得——来源广、手性体易拆分[6];稳定(相比部分含磷配合物,耐水耐氧)[7];同时四氮原子易与过渡金属螯合配位形成稳定的空间构型.在不对称氢转移研究中,形成配合物的金属离子主要采用Ru(Ⅱ)、Ir(Ⅲ)、Rh(Ⅲ)等,这些贵金属离子催化活性高,但价格昂贵,不利工业化开发和应用.若能采用价廉的第一过渡系金属代替贵金属开展不对称氢转移研究[8-9],将是很有意义的研究工作.本文以手性四氮配体和Ni(Ⅱ)形成的手性配合物为催化剂,催化苯乙酮的不对称氢转移反应,获得了较好的结果.
1 实验部分
1.1 主要仪器
所使用试剂及原料如无特殊说明,均为购置无需进一步处理.柱层析硅胶(200~300目,青岛海洋化工厂分厂);硅胶板(厚度:0.20~0.25mm,青岛海洋化工厂分厂).红外光谱仪(KBr压片,Bruker Tensor 27)、核磁共振仪(TMS为内标,Bruker公司)、熔点仪(上海精细科学仪器有限公司)、质谱仪(Agilent5975)、薄层层析用紫外灯(254nm)、气相色谱(Shimadzu GC-2014).
1.2 手性四氮配体的制备
1.2.1 (1S,2S)-N-对甲苯磺酰基-1,2-二苯基乙二胺[(S,S)-TsDPEN]的合成
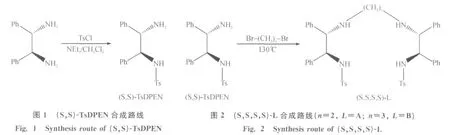
如图1按文献[10]方法合成[(S,S)-TsDPEN].在500mL的三口烧瓶中加入(S,S)-1,2-二苯基乙二胺(6.39g,30mmol)、三乙胺(13mL,93mmol)和四氢呋喃(270mL),冰水浴冷却至0~1℃.在此溶液中滴加对甲苯磺酰氯(31.4mmol)和四氢呋喃(10mL)混合液,2.5h滴加完毕,室温搅拌12h.加入40mL饱和碳酸钠溶液,用35mL二氯甲烷分液,水层二氯甲烷萃取(3×10mL),合并有机相,干燥,过滤后蒸去有机溶剂得黄色固体粉末.将此粗品溶于乙醚,加入浓盐酸(22mL),将产生的白色固体滤出后,用5%氢氧化钠洗涤,并用二氯甲烷萃取,无水硫酸钠干燥后蒸去有机溶剂得白色固体.m.p:123.2~126.9℃;[α]20589=+67.5°(c 0.4,CH2Cl2);1H NMR(CDCl3,400MHz),δ(ppm):4.399,4.386[d,2H,J=5.2Hz],4.166,4.153[d,2H,J=5.2Hz];EI-MSm/z:367[M+H]+.
1.2.2 配体A和B的合成
按文献[11]方法合成配体A和B(图2).(S,S)-TsDPEN(366mg,1.0mmol)和1,2-二溴乙烷(94mg,0.5mmol)或1,3-二溴丙烷(101mg,0.5mmol)置于密闭的玻璃仪器中,在130℃下加热28h.得到的粗产物溶解于二氯甲烷(30mL)中,用20%氢氧化钠(20mL)溶液洗涤.有机相用二氯甲烷(3× 10mL)萃取后,再用无水Na2SO4干燥,旋转蒸发得到含有四齿配体和未反应(S,S)-TsDPEN的粗产物.粗产物用硅胶柱层析法分离,得白色粉末A和B.
(A)m.p:167.5~169.2℃;[α]20589=+15.97°(c 0.4,CH2Cl2);Anal.Calcd.(%)for C44H46N4S2O4:C,69.63;H,6.11;N,7.38%;Found(%):C,69.28;H,6.14;N,7.36%.Selected IR,υ(cm-1):3 327.45,3 030.49,2 919.17,1 598.86,1 494.51,1 452.05,1 329.45,811.52,773.01,770.03,672.90.1H NMR(CDCl3,400MHz),δ(ppm):4.424,4.403[d,2H,J=8.4Hz],3.751,3.730[d,2H,J=8.4Hz],2.616,2.596[d,2H,J=8.0Hz],2.333,2.313[d,2H,J=8.0Hz],EI-MSm/z:759[M+H]+.
(B)m.p:75.2~76.8℃;[α]20589=+18.63°(c 0.4,CH2Cl2);Anal.Calcd.(%)for C45H48N4S2O4:C,69.95;H,6.22;N,7.25%;Found(%):C,70.23;H,6.48;N,6.7%.Selected IR,υ(cm-1):3 260.01,3 029.81,2 924.02,1 599.23,1 494.55,1 454.77,1 326.44,1 158.68,1 092.69,812.62,767.73,699.95,668.38.1H NMR(CDCl3,400MHz),δ(ppm):4.375,4.352[d,2H,J=7.2Hz],3.824,3.812[d,2H,J=4.8Hz],2.696[m,2H],2.561[m,2H],1.667[m,2H].EI-MSm/z:773[M+H]+.
1.3 手性四氮Ni(Ⅱ)配合物的合成
制备无水醋酸镍[12]:Ni(Ac)2·4H2O置于圆底烧瓶中,70℃下真空加热2h.得到黄绿色无水Ni(Ac)2粉末,密封备用.
将98.5mg(0.4mmol)无水Ni(Ac)2的溶于20mL无水乙醇中,得一浅绿色溶液.在此溶液中加入300mg(0.4mmol)配体A的20mL无水乙醇,N2保护下85℃回流24h,得深蓝紫色澄清溶液,旋转蒸发得紫色固体.加乙酸乙酯8mL溶解粗产物,滴加无水乙醚至大量紫色固体析出,过滤洗涤,干燥后得到:
配合物1:Ni[S,S,S,S-A](Ac)2(产率为8.2%).Anal.Calcd.(%)for C48H52N4O8S2Ni:N,6.475;C,61.540;S,6.111;H,5.747.Found(%):N,5.989;C,61.604;S,6.845;H,5.561.Selected IR,υ(cm-1):3 495.04,3 173.74,3 061.96,2 921.92,1 600.46,1 495.50,1 454.66,1 255.98,1 128.31,1 026.16,812.27,757.72,699.73,592.37.
配合物2:Ni[S,S,S,S-B](Ac)2,制备方法同上.产物为浅紫色固体(产率为7.5%).Anal.Calcd.(%)for C49H54N4O8S2Ni:N,6.051;C,61.950;S,6.693;H,5.752.Found(%):N,5.901;C,61.960;S,6.744;H,5.690.Selected IR,υ(cm-1):3 431.74,3 245.31,3 030.04,2 924.22,1 598.54,1 493.63,1 453.03,1 316.30,1 150.56,1 090.75,817.64,764.92,699.15.
配合物可能的结构如图3所示.
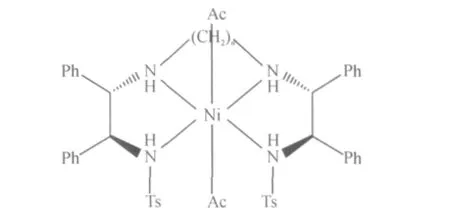
图3 推测的配合物结构(n=2,L=A;n=3,L=B)Fig.3 Proposed structure of complex 1and 2
1.4 手性Ni(Ⅱ)配合物催化苯乙酮的不对称氢转移研究[13-14]

图4 苯乙酮的不对称氢转移Fig.4 Asymmetric transfer hydrogenation of acetophenone
在氮气保护下,将0.02mmol的手性四氮Ni(Ⅱ)配合物,10mL无水异丙醇加入到50mL圆口瓶中,80℃下反应2h,用注射器注入氢氧化钾/异丙醇溶液,继续反应2~3h,再注入溶有苯乙酮(480mg,4mmol)的异丙醇溶液(S/C=100).在80℃设定温度下反应,用薄层色谱法跟踪反应进程[15].反应结束后,用2mol·L-1盐酸中和至pH=3~5,用无水乙醚萃取,合并有机相,再用饱和食盐水洗涤,无水硫酸钠干燥,过滤,浓缩,气相色谱法测定产物的转化率.配合物1和2在实验条件下催化苯乙酮不对称氢转移(图4)的转化率数据列于表1.由于转化率相对较低,不能分离得到单一型异构体,无法进行ee的测定.
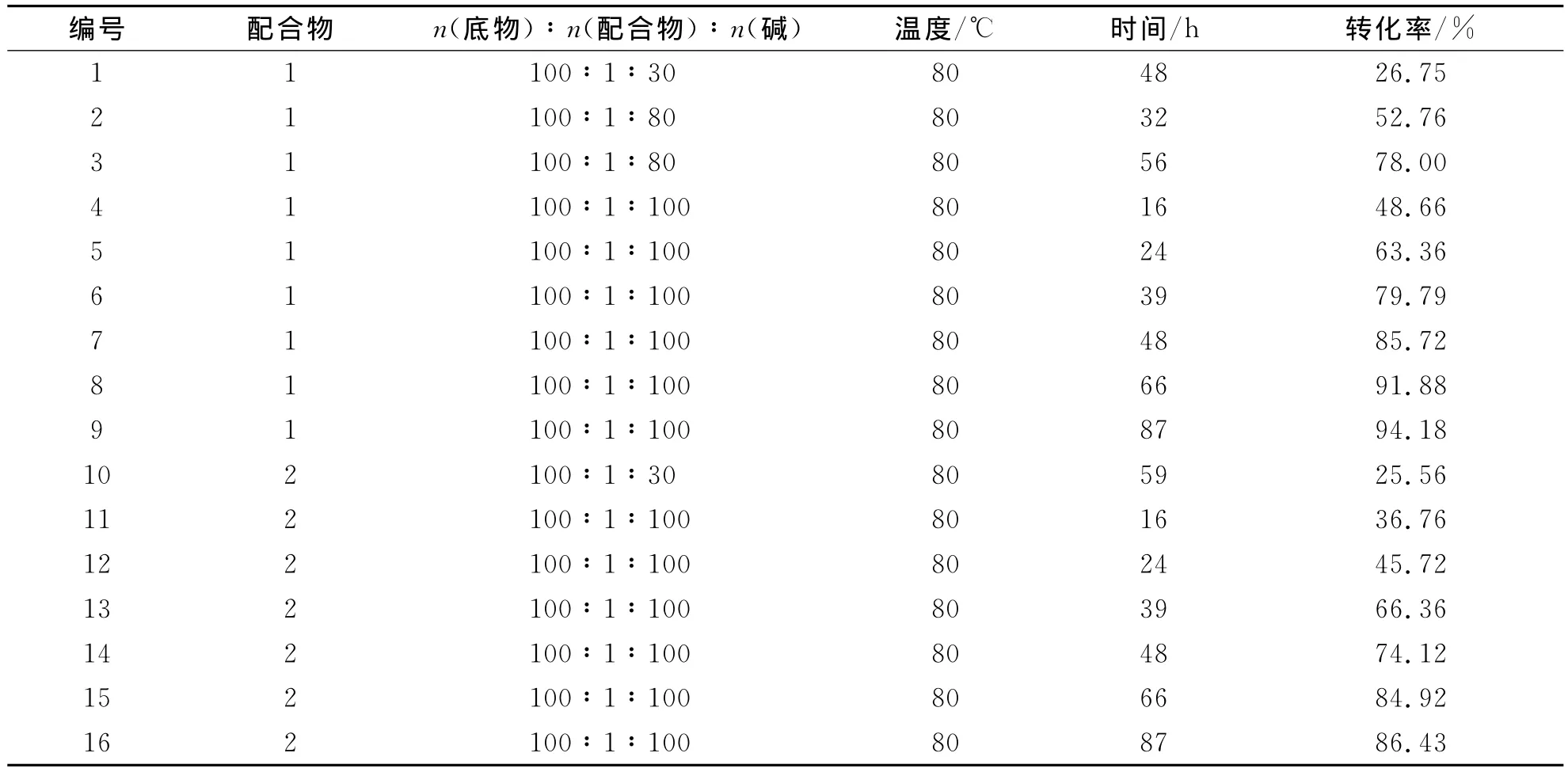
表1 配合物1和2催化苯乙酮不对称氢转移的转化率Tab.1 A comparison of conversion catalysts for ATH of acetophenone by complex 1/2
2 结果与讨论
2.1 碱量对催化活性的影响
当n(底物)∶n(碱)=1∶30时,配合物1和2催化进行48h以上转化率仍仅有30%左右(编号1和10);碱量提高至1∶80以及1∶100时,催化进行24~32h转化率即可过半(编号2和5),随着碱量的增加,配合物催化苯乙酮转化为1-苯乙醇的转化率逐渐增大.
2.2 配合物对催化活性的影响
如图5所示,固定碱量、温度等条件,反应时间在48h之内,配合物1(配体A)的催化活性优于配合物2(配体B),转化率均有明显上升;随时间延长,二者差距缩小,且增长速率趋缓.这可能是由于配合物2比配合物1多一个亚甲基,从结构上看刚性减小,导致催化效率下降.
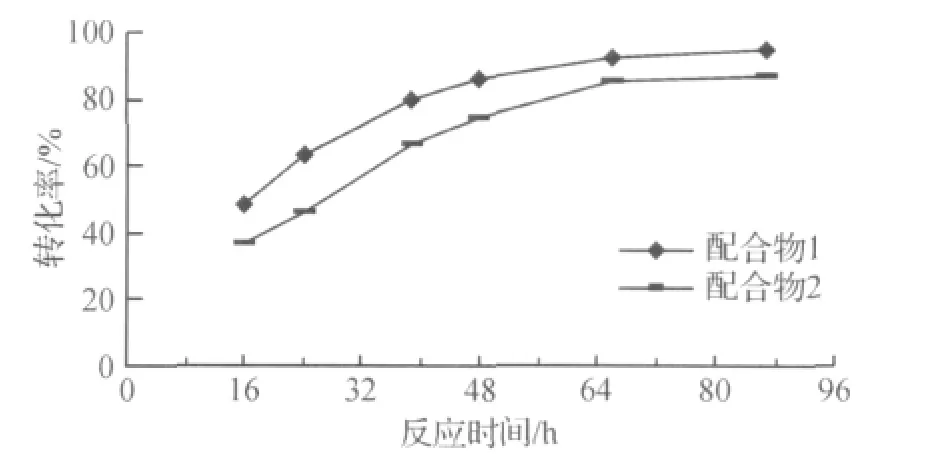
图5 碱量比100∶1时配合物1和2催化苯乙酮转化率比较Fig.5 A comparison of conversion catalysts for ATH of acetophenone by complex 1/2in 100∶1of base:complex
3 结 论
本文首次以手性四氮配体和Ni(Ⅱ)形成的手性配合物为催化剂,用于苯乙酮的不对称氢转移反应研究.该催化剂制备过程简单且价格低廉,在苯乙酮的不对称加氢中显示了一定的催化活性,在优化反应条件下,生成1-苯乙醇的转化率达到94.18%.催化剂的活性总的来说还不是很理想,有待进一步探索.
[1]Wang Chao,Wu Xiaofeng,Xiao Jianliang.Broader,green,and more efficient:Recent advances in asymmetric transfer hydrogenation[J].Chem Asian J,2008,3(10):1750-1770.
[2]林国强,王梅祥,杜灿屏,等.手性合成与手性药物[M].北京:化学化工出版社,2009:354-372.
[3]Ikariya T,Blacker A J.Asymmetric transfer hydrogenation of ketones with bifunctional transition metal-based molecular catalysts[J].Acc Chem Res,2007,40(12):1300-1308.
[4]Mao Jincheng,Guo Jun.Chiral amino amides for the ruthenium(Ⅱ)-catalyzed asymmetric transfer hydrogenation reaction of ketones in water[J].Chirality,2010,22(1):173-181.
[5]洪益玲,沈良,谭华杰.手性磺酰二胺配合物催化芳香酮的不对称氢转移研究进展[J].杭州师范大学学报:自然科学版,2010,9(1):57-64.
[6]Fonseca M H,Konig B.Chiral tetraaza ligands in asymmetric catalysis:Recent progress[J].Adv Synth Catal,2003,345(11):1173-1185.
[7]Sheldon R A.Chirotechnology:Industrial synthesis of optically active pure compounds[M].New York:Marcel Dekker,1993.
[8]杨朝芬,付海燕,熊伟,等.手性二胺修饰的镍配合物催化苯乙酮及其衍生物的不对称加氢反应[J].化学研究与应用,2007,19(4):361-365.
[9]Chen Zilu,Zeng Mulan,Zhang Yuzhen,etal.Preparation and structures of a series of phosphorus-free Nickel(Ⅱ)diamine complexes and their applications in hydrogenation of acetophenone[J].Appl Organometal Chem,2010,24(9):625-630.
[10]Xue Dong,Chen Yingchun,Cui Xin,etal.Transfer hydroge nationofactivatedC=Cbondscatalyzedbyrutheniumamidocomplexes:reaction scope,limitation,and enantioselectivity[J].J Organ Chem,2005,70(9):3584-3591.
[11]Martins J,Morris D J,Wills M.Asymmetric hydrogenation of ketones using Ir(Ⅲ)complexes of N-alkyl-N′-tosyl-1,2-ethanediamine ligands[J].Tetrahedron Lett,2009,50(6):688-692.
[12]杨瑞丽.水合醋酸镍和硫酸镍铵的热分析研究[J].咸阳师范专科学校学报:自然科学版,1998,13(6):24-26.
[13]Shen Weiyi,Zhang Hui,Zhang Hualin,etal.Novel chiral tetraaza ligands:synthesis and application in asymmetric transfer hydrogenation of ketones[J].Tetrahedron:Asymmetry,2007,18(6):729-733.
[14]Diez C,Nagel U.Chiral iridium(I)bis(NHC)complexes as catalysts for asymmetric transfer hydrogenation[J].Appl Organometal Chem,2010,24(7):509-516.
[15]He Wei,Liu Peng,Zhang Bangle,etal.Effcient iridium and rhodium-catalyzed asymmetric transfer hydrogenation using 9-amino(9-deoxy)cinchona alkaloids as chiral ligands[J].Appl Organometal Chem,2006,20(5):328-334.
Synthesis,Characterization of Ni(Ⅱ)Complexes with Chiral Tetraaza Ligands and Their Application for Asymmetric Transfer Hydrogenation of Acetophenone
ZHANG Tian,TAN Hua-jie,HONG Yi-ling,SHEN Liang
(College of Material,Chemistry and Chemical Engineering,Hangzhou Normal University,Hangzhou 310036,China)
The experiment synthesized two chiral tetraaza ligands,(S,S,S,S)-N,N′-bis(2-p-toluenesulfonylamino-1,2-diphenylethyl)ethylenediamine(A)and(S,S,S,S)-N,N′-bis(2-p-toluenesulfonylamino-1,2-diphenylethyl)trimethylenediamine(B),and characterized their Ni(II)complexes by elemental analysis,1H-NMR,MS and IR.The paper researched on the asymmetric hydrogen transfer reaction of acetophenone,and investigated the effects of the alkali charge and reaction time on ATH.
chiral tetraaza ligands;nickel complex;acetophenone;asymmetric transfer hydrogenation
O69
A
1674-232X(2012)01-0007-04
11.3969/j.issn.1674-232X.2012.01.002
2011-08-17
浙江省自然科学基金项目(Y4090056).
沈 良(1964—),男,教授,主要从事有机金属化学研究.E-mail:shenchem@hotmail.com
猜你喜欢
分子催化(2022年1期)2022-11-02
消费导刊(2019年14期)2019-08-21
消费导刊(2019年27期)2019-07-22
山东化工(2019年7期)2019-04-27
中国资源综合利用(2017年1期)2018-01-22
中国塑料(2017年2期)2017-05-17
国外医药(抗生素分册)(2016年4期)2016-07-12
国外医药(抗生素分册)(2016年2期)2016-07-12
合成化学(2015年2期)2016-01-17
苏州科技大学学报(工程技术版)(2015年3期)2015-02-28
