
硝苯地平生物黏附微球的制备及释药机制研究
2012-12-03 03:35周晓东于立军南京军区南京总医院汤山分院药械科南京211131
中国药房 2012年17期
周晓东,于立军(南京军区南京总医院汤山分院药械科,南京211131)
难溶性药物经胃肠道给药面临的难题在于如何提高其生物利用度,通过改善难溶性药物从制剂中溶出的速度和药物的溶解度,进而提高生物利用度,已成为药学工作者研究和关注的焦点[1,2]。生物黏附给药系统(Bioadhesive drug deliverysysterms,BDDS)是利用载药材料对生物黏膜表面的黏附性能,使给药系统在生物膜的特定部位滞留时间延长,或达到药物在特定部位吸收的目的[3,4]。微球(Microsphere)是指药物溶解或分散在载药材料基质中形成粒径为1~40 μm的微小球状实体,是近年来制剂研究的新热点[5,6]。本文以难溶性药物硝苯地平(Nifedipine,NFP)为模型药物,利用BDDS技术制备NFP生物黏附微球,考察其体外黏附性能和释药行为,并对其释药机制进行研究,探讨制备难溶性药物生物黏附微球的方法。
1 仪器与材料
1.1 仪器
ALC-210.4分析天平(德国Sartorius公司);752紫外分光光度计(上海精密科学仪器有限公司);ZRS-8 G型智能药物溶出仪(天津天大天发仪器有限公司);恒温水浴箱(常州国华电器有限公司)。
1.2 材料
乙基纤维素水分散体(商品名:Surelease®,上海卡乐康包衣技术有限公司);卡波姆934(简称CP,北京佳福晨康药用辅料有限公司);NFP原料药(武汉远城国际集团,批号:20100901,纯度:99.6%);NFP对照品(中国食品药品检定研究院,批号:100338,纯度:99.5%);聚乙烯醇(PVA,上海强顺化学试剂有限公司);其他试剂均为分析纯。
透析袋(天长市康鸿塑业有限公司,截留分子量:1000)。
2 方法与结果
2.1 NFP生物黏附微球的制备
取处方量的NFP溶于适量丙酮溶液中,混合均匀,磁力搅拌下将上述溶液加入分散介质司盘80中分散,搅拌1~2h。依次加入适量戊二醛(交联剂)和硫酸(固化剂),使之交联成球。减压抽去溶剂,依次用丙酮和水各洗涤3遍,取微球置于干燥器中干燥过夜。收集制得的微球,用Surelease®和CP混合液包黏附衣层,包衣温度60℃,包衣干燥时间4 h,包衣增重15%,干燥后贮存,即可得到NFP生物黏附微球;同法制备不含包衣层的微球。
2.2 NFP含量测定方法学考察
精密称取干燥至恒重的NFP 30 mg,用少许甲醇溶解后再以乙醇定容至100 mL,作为贮备液。精密吸取贮备液1、2、3、4、5、6mL置于50mL棕色量瓶中,以0.5%十二烷基硫酸钠(SDS)水溶液定容至刻度,制备成硝苯地平浓度分别为6、12、18、24、30、36 μg·mL-1的系列标准溶液;以0.5%SDS水溶液为空白,在333 nm波长下测定吸光度。以浓度(x)为横坐标,吸光度(y)为纵坐标,进行线性回归处理,得回归方程为y=0.0115 x+0.0015(r=0.9993),结果,NFP检测浓度线性范围为6~36 μg·mL-1。分别进行低、中、高浓度(10、20、30 μg·mL-1)的回收率试验和精密度试验,结果低、中、高浓度的平均回收率为99.5%(RSD=1.8%),日内RSD分别为2.3%、1.5%、2.2%,日间RSD分别为2.4%、1.8%、2.0%,表明方法的准确度和精密度均符合要求。
2.3 正交试验设计优化生物黏附微球处方工艺
在前期单因素考察的基础上,设计制备微球,总处方量为5mL,分别对影响微球质量的因素进行考察:加热温度(A,℃)、PVA用量(B,g)、司盘浓度(C,%)、戊二醛用量(D,mL)。每个因素选取3个水平,选用L9(34)正交设计表,以药物24 h累积释药率为评价指标,优化微球处方。L9(34)正交试验设计因素和水平见表1,正交试验结果见表2,方差分析见表3。

表1 因素和水平Tab 1 Factor and levels
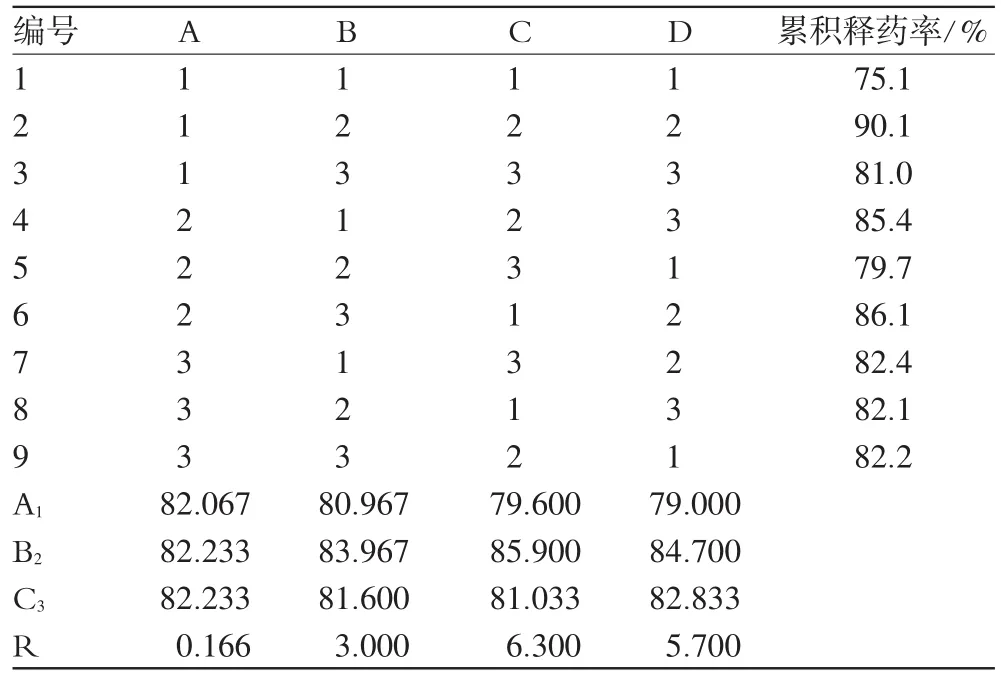
表2 正交试验结果Tab 2 Design and results of orthogonal design
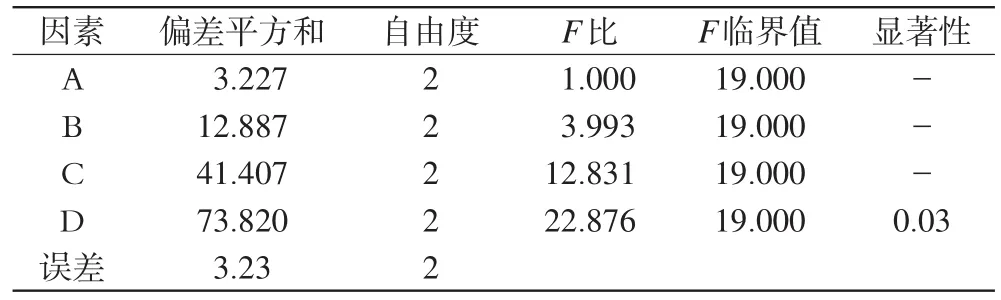
表3 方差分析结果Tab 3 Result of variance analysis
由直观分析可知,各因素对药物释放的影响大小顺序为因素C>D>B>A。通过方差分析可知,D因素的变化有统计学差异,故优选D2,而A、B、C因素的变化无统计学差异,综合考虑,确定处方优选结果为D2A2B2C2,即加热温度为60℃,PVA用量为0.6 g,司盘浓度为5%,戊二醛用量为0.6 mL。以此制备3批样品,进行质量考察。
2.4 微球的质量考察
2.4.1 载药量、包封率的测定。取微球加入研钵中研磨,精密称量50 mg,置于25 mL量瓶中,加入适量0.5%SDS溶液后,超声溶解,定容,摇匀,静置。滤过,取续滤液备用。精密量取续滤液1 mL置于100 mL量瓶中,加入0.5%SDS溶液稀释至刻度,摇匀。取以相同方法制备的空白微球溶液和对照品溶液,于333 nm波长处测定吸光度。每批微球测定3次,由外标法计算药物浓度,根据下列公式计算微球包封率和载药量[7]:包封率(%)=(微球中的含药量/微球与介质中的总药量)×100%,载药量(%)=(微球中NFP质量/微球总质量)×100%。结果平均载药量约为15%,包封率约为85%,详见表4。
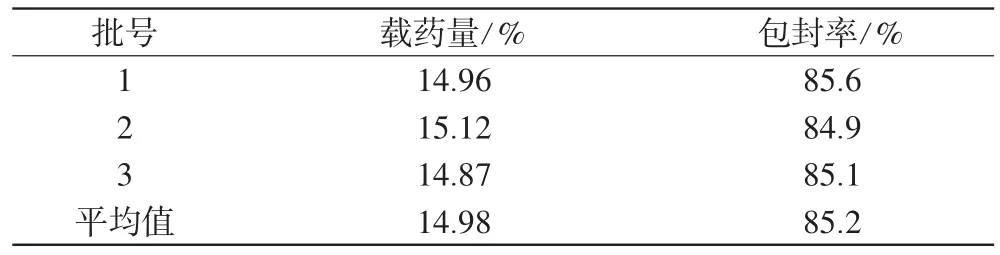
表4 3批微球载药量及包封率测定结果Tab 4 Results of drug-loading amount and encapsulation efficiency of 3batches of microspheres
2.4.2 微球粒径的测定。取微球适量,分散于适量水中,超声震荡15 min后,均匀涂布于载玻片上,采用光学显微观察微球的粒径及分布。根据公式D=∑(nd)/∑n(D:平均粒径;n:微球个数;d:统计范围内微球等价粒径的平均值)计算800个微球的粒径得平均粒径,再根据800个微球粒径的统计结果,分析微球粒径的分布范围[7],结果见图1。

图1 微球粒径分布图Fig 1 The particle size distribution of microspheres
从图1可见,本法制备的微球平均粒径约为20µm,其粒径85%均匀分布在16~20µm左右,可知粒径分布均匀。
2.4.3 体外黏附性能的评价。采用自制的体外黏附力测定装置[8]。试验时用502胶水将适量生物黏附微球的一侧固定于2 cm×2 cm背面带挂钩的聚氯乙烯薄片B上,备用;将已准备的大鼠胃组织剪成小片,用502胶水固定于相同规格的薄片A上。滴加生理盐水5滴于微球表面润湿10 min后与固定于薄片A的胃组织接触,并给予500 g压力,施压时间5 min,将薄片A挂钩垂直固定,在薄片B挂钩上挂系一塑料袋,以8~10 mL·min-1的速度向袋中注水,直至微球下侧与黏膜分离,记录塑料袋、水及薄片B的重量(g),即得体外最大黏附力。测定3批微球的最大体外黏附力并与未包衣的微球比较,结果见表5。

表5 2种微球体外黏附力测定结果Tab 5 The adhesive ability of 2kinds of microspheres in vitro
由表5可知,黏附微球体外黏附力明显高于未包衣微球,前者具有较好的黏附效果。
2.4.4 体外释放性考察。以900 mL 0.5%SDS溶液为释放介质,保持温度在(37±1)℃。精密称取适量微球,置于透析袋中,将透析袋放入释放杯中,开启搅拌(100 r·min-1)。分别于0、1、2、4、8、12、14、18、24 h取样5 mL(同时在溶出杯中补入相同体积的新鲜释放介质)。滤过,精密量取续滤液1 mL,置于25 mL量瓶中,加入0.5%SDS溶液至刻度,摇匀。取相同方法制备的空白微球溶液和对照品溶液,于333 nm波长处测定吸光度。按外标法计算各点的药物浓度,求算3批微球的累积释药率,绘制体外释药曲线,见图2。

图2 微球的体外释药曲线(n=3)Fig 2 Drug release curves of microspheres in vitro(n=3)
由图2可见,微球可持续24 h释药;体外释药曲线分别按零级、一级、Higuchi方程拟合,结果显示所制微球数据符合Higuchi释药动力学方程(r>0.9900),具有较好的体外缓释性能。
2.5 释药机制研究
根据药物从骨架中释放机制的Ritger-Peppas方程[9]可知:Mt/M∞=ktn。式中:Mt/M∞为药物在某一时间的累积释药率;t为释放时间;k为常数,该常数随不同药物或不同处方以及不同释放条件而变化,表示释放速率大小的重要参数;n为释放参数,为Ritger-Peppas方程中表示释放机制的特征参数(当n<0.45时,服从Fick扩散;当n>0.89时,为骨架溶蚀机制;当0.45<n<0.89时,为非Fick扩散机制,药物释放机制为混合型,即药物释放为药物的扩散和骨架溶蚀双重机制[9])。
结果,Ritger-Peppas拟合方程为:lnQ=0.7512 t+2.0821(r=0.9978),其中n=0.7512(0.45<n<0.85),为非Fick扩散机制,表明NFP从自制黏附微球中以扩散和骨架溶蚀2种途径释放。
3 讨论
本试验中采用乙基纤维素水分散体Surelease®和CP混合作为外层黏附衣层,一方面可以通过Surelease®适当的膜孔有效调节NFP的释放行为,另一方面可以借助CP的黏附性能使微球较长时间地黏附于胃肠黏膜[10],增加药物吸收,以期提高难溶性药物的生物利用度。
生物黏附微球的制备过程中可变因素较多,本文在单因素考察的基础上通过正交设计优化处方工艺,分别考察了加热温度等各因素对微球释药的影响,最终以优选处方工艺连续制备3批样品并评价其质量。结果显示所制微球具有缓释性,其释药机制符合非Fick扩散机制,药物以溶蚀和扩散2种模式释放。
本研究建立的体外黏附力的测定可在一定程度上反映制剂体内的黏附性能,但准确度仍需进一步探明。同时,NFP生物黏附微球体内释药行为及体内吸收的相关性等仍有待于进一步研究。
[1]Pinnamaneni S,Das NG,Daa SK.Formulation approaches for orally administered poorly soluble drugs[J].Pharmazie,2002,57(5):291.
[2]白志华,方晓玲.难溶性药物的口服制剂研究进展[J].中国药学杂志,2005,40(15):1124.
[3]林文慧.葛根总黄酮胃肠道生物黏附制剂的研究[D].北京:北京协和医学院,2008:12.
[4]盛 丰.生物黏附给药系统研究概述[J].中国医学工程,2010,18(2):180.
[5]龚 伟,杨 阳,金义光,等.新型药物递送系统研究进展[J].生命科学,2011,41(10):894.
[6]唐振香,苗青原.药物靶向制剂的研究进展[J].中国药事,2007,21(8):628.
[7]Chen AZ,Li Y,Chen D.Development of core-shell microcapsules by a novel supercritical CO2process[J].Mater Sci Mater Med,2009,20(3):751.
[8]赵振霞.格列吡嗪胃肠道生物黏附缓释片的研制[D].石家庄:河北医科大学,2009:5.
[9]任君刚.NFP缓释微丸的研究[D].沈阳:沈阳药科大学,2006:10.
[10]卢 毅,张 彤,陶建生.卡波姆在药剂学中的应用[J].中国医药工业杂志,2007,38(6):457.
猜你喜欢
环境卫生工程(2021年4期)2021-10-13
婚姻与家庭·婚姻情感版(2021年6期)2021-06-01
潍坊学院学报(2020年6期)2020-11-22
化工科技(2020年1期)2020-03-16
中国兽医杂志(2018年9期)2018-12-29
中成药(2018年9期)2018-10-09
中成药(2017年5期)2017-06-13
中成药(2017年6期)2017-06-13
海峡科技与产业(2016年3期)2016-05-17
小说月刊(2015年6期)2015-12-16
