
siRNA沉默RALA基因影响人白血病K562细胞增殖和凋亡*
2012-11-06 06:06:34李育敏朱雪姣谷景义
中国病理生理杂志 2012年4期
李育敏, 朱雪姣, 谷景义, 费 嘉
(暨南大学医学院生物化学与分子生物学教研室, 广东 广州 510632)
1000-4718(2012)04-0625-06
2011-10-08
2012-02-24
国家自然科学基金资助项目(No.81170496);暨南大学科研培育与创新基金(前瞻性与基础)研究项目(No.21609406)
△通讯作者 Tel: 020-85220256; E-mail: efeijia@163.com
siRNA沉默RALA基因影响人白血病K562细胞增殖和凋亡*
李育敏, 朱雪姣, 谷景义, 费 嘉△
(暨南大学医学院生物化学与分子生物学教研室, 广东 广州 510632)
目的研究小干扰RNA(siRNA)抑制v-ral 猴白血病病毒癌基因同系物A(RALA)基因表达对人慢性粒细胞性白血病K562细胞增殖和凋亡的影响。方法利用LipofectamineTM2000将化学合成的RALA siRNA转染体外培养的K562细胞,四甲基偶氮唑蓝(MTT)法检测RALA siRNA对K562细胞增殖的影响;台盼蓝拒染法检测RALA siRNA对K562细胞存活率的影响;real-time PCR检测RALA mRNA表达水平;Western blotting检测RALA蛋白表达水平;annexin V/PI双染流式细胞仪检测RALA siRNA对K562细胞凋亡的影响;Hoechst 33258染色荧光显微镜观察细胞形态学变化。结果RALA siRNA可明显抑制K562细胞内RALA mRNA和蛋白的表达(P<0.05);与阴性对照组相比,转染RALA siRNA的K562细胞增殖明显受到抑制(P<0.05);流式细胞术结果显示转染RALA siRNA的K562细胞凋亡较阴性对照组显著增加(P<0.05);Hoechst 33258染色见转染RALA siRNA的K562细胞出现典型的凋亡形态学变化。结论癌基因RALA在白血病发生发展过程中发挥重要作用。siRNA下调RALA mRNA和蛋白的表达,可抑制人白血病K562细胞增殖,诱导细胞凋亡,提示RALA可能是白血病治疗的新靶点。
v-ral猴白血病病毒癌基因同系物A; 小干扰RNA; 白血病; 细胞增殖; 细胞凋亡
慢性粒细胞白血病(chronic myelogenous leukemia, CML)是慢性骨髓增生性疾病中最常见的一种,属于造血干细胞恶性增殖性疾病,以粒系增生为主。90% CML病人都存在BCR-ABL融合蛋白[1]。近年来的研究表明BCR-ABLl融合蛋白与Ras信号通路之间存在着密切的关系,Ras超家族成员如Rac、CDC42等在BCR/ABL融合蛋白下游作用通路中扮演着重要角色,提示Ras信号通路与CML发病机制密切相关[2-4]。v-ral猴白血病病毒癌基因同系物A(v-ral simian leukemia viral oncogene homolog A,RALA)是Ral(Ras-like)家族的一员,其翻译的蛋白质类似于ras基因翻译的连接于三磷酸鸟苷(guanosine triphosphate, GTP)上的连接蛋白,这种蛋白可以与细胞膜上的受体结合,调节细胞内外物质的转运。研究表明Ral家族成员与肿瘤发生发展密切相关,在肺癌、结肠癌和胰腺癌等肿瘤中都发现Ral家族基因的活化、突变和表达改变[5-7]。RALA作为一种癌基因,参与多种肿瘤细胞增殖、凋亡、转移和侵袭等一系列进程[8-10]。而RALA是否参与CML的发生发展尚待研究。RNA干扰(RNA interference,RNAi)现象自发现以来,已成功应用于线虫、果蝇、植物、真菌和哺乳动物等生物的多个研究领域中[11]。本实验采用RNAi技术抑制癌基因RALA的表达,观察其对人CML细胞株K562细胞增殖和凋亡的影响,研究RALA在K562细胞增殖和凋亡过程中的作用机制,为研究CML新的治疗靶点提供实验依据。
材 料 和 方 法
1材料
人慢性粒细胞白血病K562细胞株购于中国科学院上海细胞库。RALA siRNA和随机序列由上海吉玛制药技术有限公司设计合成,-20 ℃保存。RALA siRNA正义链引物5′-CGUGGAAACAUCUGCUAAATT-3′, 反义链引物5′-UUUAGCAGAUGUUUCCACGTA-3′;随机序列(scramble, SCR)正义链引物5′-UUCUCCGAACGUGUC ACGUTT-3′, 反义链引物5′-ACGUGACACGUUCGGAGAATT -3′。DMEM液体培养基和胎牛血清购自Gibco。Opti-MEM培养基购自Invitrogen。LipofectamineTM2000 购自Invitrogen。MTT、台盼蓝、二甲基亚砜(DMSO)和Hoechst 33258试剂均购自Sigma。Trizol试剂和real-time PCR试剂购自北京天根生化科技有限公司。RIPA细胞裂解液购自上海申能博彩生物科技有限公司。BCA蛋白浓度测定试剂盒购自北京博奥森生物技术有限公司。鼠抗β-actin多抗购自武汉博士德生物工程有限公司。兔抗RALA多抗购自Abcam。ECL试剂盒购自碧云天生物技术研究所。
2方法
2.1细胞培养 将K562细胞接种于含体积分数10%的胎牛血清、无抗生素的DMEM/F12培养基中,置于37 ℃、体积分数为5%的CO2培养箱,饱和湿度下培养。每2~3 d换液传代。实验选用对数生长期、0.2%台盼蓝拒染率>95%的细胞。
2.2MTT法测细胞增殖抑制率 实验分RALA siRNA组、随机对照组(随机序列组,阴性对照)和空白对照组,每组设4个复孔。RALA siRNA组和随机对照组的核酸终浓度为25、50、75、100、125 nmol/L。取对数生长期细胞,各组细胞以1×108/L的密度接种于96孔板,每孔50 μL,转染体积100 μL(转染方法参照LipofectamineTM2000说明书),空白对照组加入与药物等体积的无血清Opti-MEM培养基。转染6 h后,加入100 μL含20%血清DMEM/F12培养基,使终体积200 μL。48 h后每孔加入MTT液20 μL,于培养箱中培养4 h后,1 000 r/min离心10 min,弃上清,每孔加入150 μL DMSO。振荡使结晶充分溶解,在多功能酶标仪上测定吸光度A570,间接反映存活细胞量。实验重复3次,计算增殖抑制率。增殖抑制率(%)=[1-(A实验组/A对照组)]×100%。
2.3台盼蓝拒染法检测不同时段的细胞生长状况 实验分组及转染处理同前,RALA siRNA组和随机对照组的核酸终浓度为100 nmol/L。于24、48、72 h台盼蓝拒染法检测各组活细胞数。实验重复3次,取均值。
2.4Real-time PCR检测细胞内RALA mRNA的相对表达水平 实验分组同前。取对数生长期细胞,各组细胞以1.5×108/L的密度接种于24孔板,每孔400 μL,转染终体积500 μL。6 h后加入500 μL 20%血清培养基置于培养箱中培养。RALA siRNA组和随机对照组的核酸终浓度为100 nmol/L。于48 h收集细胞,总RNA抽提按Trizol试剂说明书进行,提取的总RNA用紫外分光光度仪(UVP)测A260/A280进行RNA纯度及浓度计算, 利用SYBR GreenⅠreal-time PCR检测K562细胞内RALA mRNA的表达水平,以GAPDH为内参照。逆转录条件:37 ℃持续1 h, 95 ℃持续5 min。Real-time PCR条件:94 ℃ 5 min,94 ℃ 30s,RALA 56 ℃或GAPDH 57.5 ℃ 30 s,72 ℃ 30 s,重复循环40次, 融解曲线分析60 ℃~95 ℃。PCR所用引物自行设计,由上海生物工程技术有限公司合成,RALA上游引物5′-ATCGGAAGAAGGTAGTGC-3′,下游引物5′-AAATCTGCTCC CTGAAGT-3′;GAPDH上游引物5′-CAACGGATTTGGTCGTATT-3′,下游引物5′-CACAGTCTTCTGGGTGGC-3′。RALA和GAPDH的扩增片段长度分别为182 bp和541 bp。RALA mRNA的相对表达水平用ΔCt和2-ΔΔCt计算。
2.5Western blotting检测细胞内RALA蛋白的相对表达水平 实验分组及转染方法同前。转染48 h后,离心收集细胞,按细胞裂解液(RIPA)说明书提取细胞蛋白。用BCA蛋白定量试剂盒进行蛋白定量,根据定量结果对各蛋白质样本进行校正。加入样品溶解液和样品,置沸水中煮 5 min。不连续的聚丙烯酰胺凝胶电泳(10%聚丙烯酰胺分离胶和5%聚丙烯酰胺浓缩胶),电压80 V,进入分离胶后改为120 V,40 min。将凝胶上的蛋白条带转到硝酸纤维膜上,半干式转膜15 V,18 min;TBST洗膜5 min。5%牛血清白蛋白封闭缓冲液室温封闭1 h。TBST洗膜3次,每次5 min。4 ℃过夜孵育Ⅰ抗。TBST洗膜3次,每次5 min。37 ℃孵育Ⅱ抗1 h。TBST洗膜3次,每次5 min。加入ECL,显影、定影、扫描。BI-2000型图像分析软件分析RALA和β-actin的积分吸光度,以β-actin作为内参照计算RALA蛋白的相对表达水平。
2.6Annexin V/PI双染检测RALA siRNA对细胞凋亡的影响 实验分组及转染处理同前,转染后48 h离心收集各组细胞,预冷PBS洗涤细胞3次,1 mL结合缓冲液洗细胞1次,离心去上清,加入200 μL结合缓冲液,重悬细胞后,分别加入10 μL annexin V和5 μL PI,轻轻混匀,避光反应30 min,立即上机检测。Annexin V(+)、PI(-)的细胞为早期凋亡细胞。Annexin V(+)、PI(+)的细胞为晚期凋亡细胞和坏死细胞。
2.7Hoechst 33258染色观察凋亡细胞形态学变化 实验分组及转染处理同前,转染后48 h离心收集各组细胞,预冷PBS洗涤细胞3次,留少许上清,用微量加样器吸取细胞在洁净载玻片上涂片,室温自然干燥,甲醇∶冰醋酸(3∶1)固定液固定10 min,Hoechst 33258 染色工作液(10 mg/L)避光染色2 min,流水冲洗5 min,荧光显微镜观察并拍照。
3统计学处理
结 果
1MTT法检测RALAsiRNA对K562细胞增殖影响的量效效应
RALA siRNA转染K562细胞48 h后,表现出对细胞增殖的抑制作用。25、50、75、100和125 nmol/L RALA siRNA组与对照组相比,均有统计学意义(P<0.05)。100 nmol/L RALA siRNA对细胞增殖的抑制作用最佳,达125 nmol/L浓度时,出现对细胞的非特异性抑制作用。随着浓度的增加,RALA siRNA对细胞增殖的抑制作用越明显,呈剂量依赖关系,见图1。

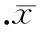
图1RALAsiRNA抑制K562细胞增殖的量效关系
2台盼蓝拒染法检测RALAsiRNA对K562细胞存活率影响的时间效应
100 nmol/L的RALA siRNA作用于K562细胞后24 h开始表现出抑制细胞存活率的效应,持续至72 h,与对照组相比有显著差异(P<0.05),见图2。
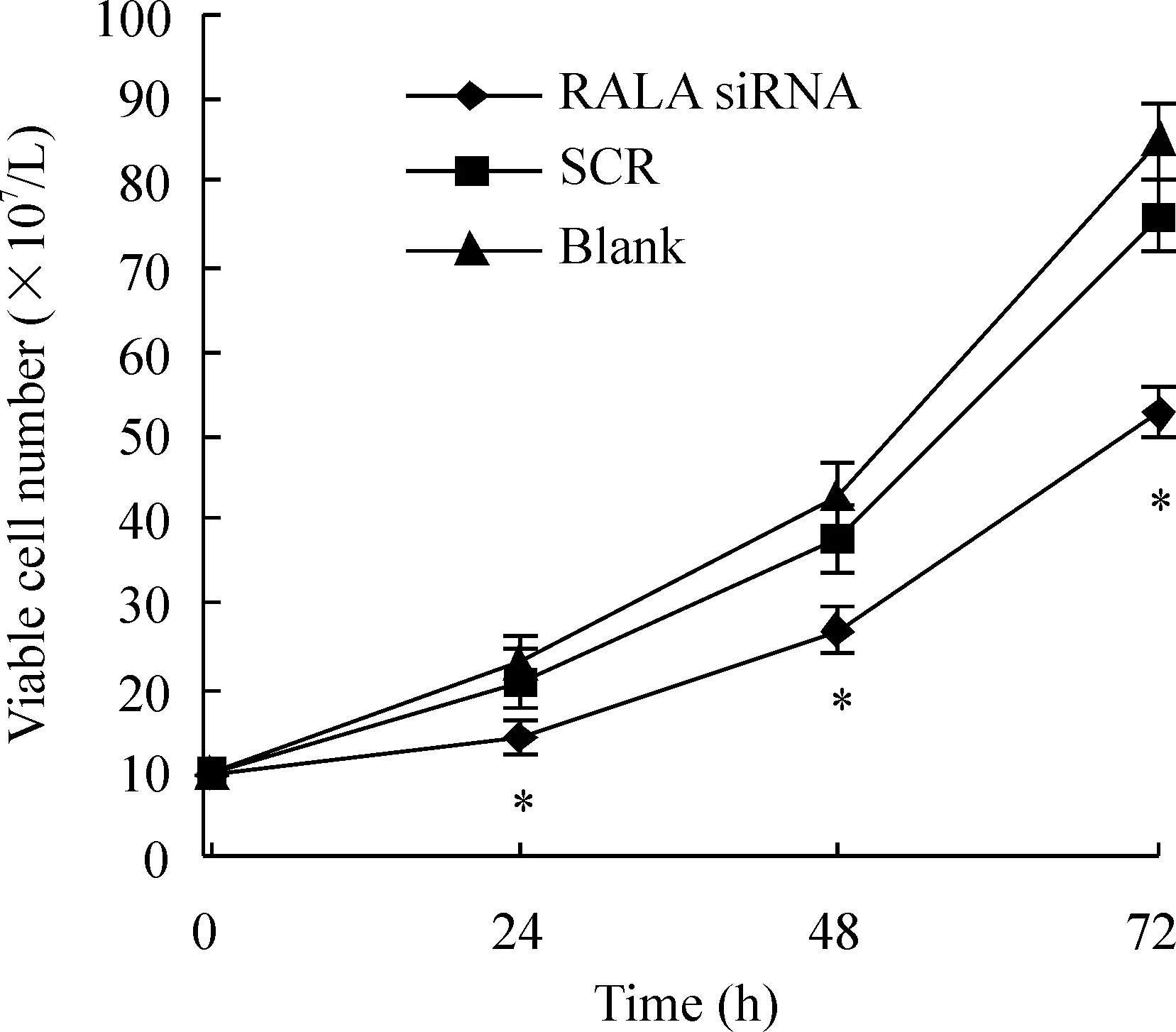
图2RALAsiRNA对K562细胞存活率的影响
3Real-timePCR检测RALAsiRNA对K562细胞内RALAmRNA的表达水平影响
Real-time PCR结果显示:RALA siRNA转染K562细胞48 h后,与对照组相比,RALA siRNA组RALA mRNA的表达量显著降低,差异有统计学意义(P<0.05),见图3。

图3Real-timePCR检测K562细胞RALAmRNA的表达
4Westernblotting检测RALAsiRNA对K562细胞内RALA蛋白表达的影响
RALA siRNA转染K562细胞48 h后,Western blotting检测RALA蛋白的表达。结果显示,与对照组相比,RALA siRNA组RALA蛋白的表达量显著降低,差异有统计学意义(P<0.05),见图4。
5AnnexinV/PI双染检测RALAsiRNA对K562细胞凋亡的影响
空白对照组和随机对照组细胞早期凋亡率分别为2.54%±0.87%和2.85%±1.04%,RALA siRNA组细胞出现早期凋亡细胞群,早期凋亡率为7.74%±0.72%,与随机对照组相比,差异有统计学意义(P< 0.05),见图5。

图4Westernblotting检测K562细胞RALA蛋白的表达
6Hoechst33258染色观察K562细胞形态学变化
随机对照组细胞与空白对照组相比未见明显形态学差异,RALA siRNA组细胞可见核固缩、边聚、裂解和凋亡小体形成等形态学变化,见图6。

图5AnnexinV/PI双染检测RALAsiRNA对K562细胞凋亡的影响

Figure 6. The morphological character of K562 cells transfected with RALA siRNA(Hoechst 33258 staining,×40)
图6K562细胞形态学变化
讨 论
CML是一种起源于多能造血干细胞的恶性增殖性疾病,约占成年人白血病的20%~30%,在我国白血病中发病率仅次于急性淋巴细胞性白血病和急性髓系白血病而居第3位,成为威胁人民健康的一种常见恶性肿瘤。目前治疗CML的常规化疗药物如马利兰和羟基脲等缺乏肿瘤特异性,常引起较大的毒副作用。异基因造血干细胞移植是当今唯一能治愈CML的方法,但受年龄、供者、病程、经济条件和移植相关病等多种因素限制,仅少数患者有条件接受。近年来基因靶向药物——甲磺酸伊马替尼(imatinib mesylate)已成为CML治疗的一线药物,但随之产生的耐药性往往导致治疗失败,而且其价格极为昂贵。因此,寻求肿瘤恶性转化的关键“靶标”以及与肿瘤发生密切相关的基因,开发新的靶向药物,仍是肿瘤基因治疗研究的重要方向。
RALA蛋白的功能依赖于活化型与失活型Ral之间的循环转化能力,即Ral磷酸化和去磷酸化的能力,受Ral特异的鸟嘌呤核苷酸交换因子(Ral-specific guanyl nucleotide exchange factor,RalGEF)调节。RalGEF是Ras致细胞癌变的关键因素,与癌症的发生发展密切相关[9, 12-13]。慢性粒细胞性白血病的Ph染色体形成BCR-ABL融合基因,产生一种具有酪氨酸激酶活性的BCR-ABL融合蛋白,从而促发CML。近年来研究表明Ras信号通路在BCR-ABL融合蛋白下游基因调控作用中扮演着重要角色,与CML发病机制密切相关[2-4]。本实验设计合成RALA siRNA,转染人慢性粒细胞性白血病细胞株K562细胞,结果显示RALA siRNA显著下调K562细胞内RALA mRNA和蛋白的表达,抑制细胞增殖(P<0.05)。转染RALA siRNA的K562细胞早期凋亡率较随机对照组增高(P<0.05),可见核固缩、边聚、裂解、凋亡小体形成等凋亡形态学变化。这表明在K562细胞中癌基因RALA的表达水平与细胞增殖和凋亡密切相关,RALA可能是白血病基因治疗的新靶点。
研究表明,癌基因RALA通过调控一系列下游分子参与肿瘤的发生发展机制。Ral相互作用蛋白(Ral-interacting protein, RLIP)76[又称RALA结合蛋白1(RALA-binding protein 1,RALBP1)]是 RALA的效应分子[14]。肿瘤坏死因子相关的凋亡诱导配体(tumour necrosis factor-related apoptosis-inducing ligand,TRAIL),又称肿瘤坏死因子超家族成员10(tumor necrosis factor superfamily, member 10, TNFSF10),参与RALA-RLIP途径,抑制抗凋亡蛋白caspase-8(CASP8)和CASP8与Fas相关死亡域蛋白样凋亡调控因子(CASP8 and FADD-like apoptosis regulator, CFLAR)的翻译[15]。c-Jun 氨基端激酶1(c-Jun N-terminal kinase 1, JNK1)[又称丝裂原活化蛋白激酶8(mitogen-activated protein kinase 8,MAPK8)]和核因子(nuclear factor κB,NF-κB)也参与调节RLIP[16-19]。RLIP的表达降低使肿瘤细胞生长和存活受抑制[20-21]。CD24也是RALA的下游效应分子,CD24参与调节细胞凋亡[22]。Ras 1激酶抑制因子连接体增强子(connector enhancer of kinase suppressor of Ras 1, CNK1)与RALA相互干扰,CNK1促进凋亡[23]。研究提示,抑制PI3K/Akt信号通路可抑制肿瘤细胞的增殖并诱导凋亡的发生[24]。散射因子(scatter factor,SF)是一种肝细胞生长因子,它在肿瘤细胞内大量堆积,对抗DNA损伤剂引起的细胞毒性和凋亡,从而对肿瘤细胞起到保护作用。SF与受体(c-Met) 结合,通过 PI3K → c-Akt → PAK1(p21-activated kinase 1) → NF-κB通路发挥SF介导的细胞保护和抗凋亡机制,RALA参与调节SF的这种功能[25],而PI3K/Akt通路激活可抑制细胞凋亡[26],因此RALA可能通过该途径参与调节细胞凋亡。最近研究表明,RALA参与调节细胞有丝分裂过程中线粒体的分布,从而影响细胞的分裂和增殖[27]。在本实验中,我们还没有对RALA影响细胞增殖和凋亡的下游分子机制进行深入的研究。我们推测在K562细胞中,RALA也可能通过调控这些下游分子来影响肿瘤细胞的增殖和凋亡。这些已经取得的研究成果为我们今后的研究工作指明了方向。作为Ral家族成员的RALA与BCR-ABL融合蛋白的关系将是我们下一步深入探讨的问题。
[1] Koschmieder S, Gottgens B, Zhang P, et al. Inducible chronic phase of myeloid leukemia with expansion of hematopoietic stem cells in a transgenic model of BCR-ABL leukemogenesis[J]. Blood, 2005, 105(1):324-334.
[2] Puil L, Liu J, Gish G, et al. Bcr-Abl oncoproteins bind directly to activators of the Ras signalling pathway[J]. EMBO J, 1994, 13(4):764-773.
[3] Thomas EK, Cancelas JA, Chae HD, et al. Rac guanosine triphosphatases represent integrating molecular therapeutic targets for BCR-ABL-induced myeloproliferative disease [J]. Cancer Cell, 2007, 12(5):467-478.
[4] Harnois T, Constantin B, Rioux A, et al. Differential interaction and activation of Rho family GTPases by p210bcr-abland p190bcr-abl[J]. Oncogene, 2003, 22(41):6445-6454.
[5] Knizhnik AV, Kovaleva OB, Laktionov KK, et al. Arf6, RalA and BIRC5 protein expression in non small cell lung cancer [J]. Mol Biol(Mosk), 2011, 45(2):307-315.
[6] Martin TD, Samuel JC, Routh ED, et al. Activation and involvement of Ral GTPases in colorectal cancer[J]. Cancer Res, 2011, 71(1):206-215.
[7] Lim KH, O’Hayer K, Adam SJ, et al. Divergent roles for RalA and RalB in malignant growth of human pancreatic carcinoma cells[J]. Curr Biol, 2006, 16(24):2385-2394.
[8] Lim KH, Baines AT, Fiordalisi JJ, et al. Activation of RalA is critical for Ras-induced tumorigenesis of human cells[J]. Cancer Cell, 2005, 7(6):533-545.
[9] Bodemann BO, White MA. Ral GTPases and cancer: linchpin support of the tumorigenic platform[J]. Nat Rev Cancer, 2008, 8(2):133-140.
[10]Chien Y, White MA. RAL GTPases are linchpin modulators of human tumour-cell proliferation and survival[J]. EMBO Rep, 2003, 4(8):800-806.
[11]Fire A, Xu SQ, Montgomery MK, et al. Potentand specific genetic interference by double-stranded RNA inCaenorhabditiselegans[J]. Nature, 1998, 391(6669):806-811.
[12]Hamad NM, Elconin JH, Karnoub AE, et al. Distinct requirements for Ras oncogenesis in human versus mouse cells[J]. Genes Dev, 2002, 16(16): 2045-2057.
[13]Rangarajan A, Hong SJ, Gifford A, et al. Species- and cell type-specific requirements for cellular transformation[J]. Cancer Cell, 2004, 6(2): 171-183.
[14]Cantor SB, Urano T,Feig LA. Identification and characterization of Ral-binding protein 1, a potential downstream target of Ral GTPases[J]. Mol Cell Biol, 1995, 15(8): 4578-4584.
[15]Panner A, Nakamura JL, Parsa AT, et al. mTOR-independent translational control of the extrinsic cell death pathway by RalA[J]. Mol Cell Biol, 2006, 26(20): 7345-7357.
[16]Henry DO, Moskalenko SA, Kaur KJ, et al. Ral GTPases contribute to regulation of cyclin D1 through activation of NF-κB[J]. Mol Cell Biol, 2000, 20(21): 8084-8092.
[17]de Ruiter ND, Wolthuis RM, van Dam H, et al. Ras-dependent regulation of c-Jun phosphorylation is mediated by the Ral guanine nucleotide exchange factor-Ral pathway[J]. Mol Cell Biol, 2000, 20(22): 8480-8488.
[18]Micheau O, Lens S, Gaide O, et al. NF-κB signals induce the expression of c-FLIP[J]. Mol Cell Biol, 2001, 21(16): 5299-5305.
[19]Chang L, Kamata H, Solinas G, et al. The E3 ubiquitin ligase itch couples JNK activation to TNFα-induced cell death by inducing c-FLIPLturnover[J]. Cell, 2006, 124(3): 601-613.
[20]Singhal SS, Awasthi YC, Awasthi S. Regression of melanoma in a murine model by RLIP76 depletion[J]. Cancer Res, 2006, 66(4): 2354-2360.
[21]Singhal SS, Singhal J, Yadav S, et al. Regression of lung and colon cancer xenografts by depleting or inhibiting RLIP76(Ral-binding protein 1)[J]. Cancer Res, 2007, 67(9): 4382-4389.
[22]Smith SC, Oxford G, Wu Z, et al. The metastasis-associated geneCD24 is regulated by Ral GTPase and is a mediator of cell proliferation and survival in human cancer[J]. Cancer Res, 2006, 66(4): 1917-1922.
[23]Rabizadeh S, Xavier RJ, Ishiguro K, et al. The scaffold protein CNK1 interacts with the tumor suppressor RASSF1A and augments RASSF1A-induced cell death[J]. J Biol Chem, 2004, 279(28): 29247-29254.
[24]费洪荣, 陈洪蕾, 辛晓明, 等. Akt抑制剂perifosine抑制胃癌细胞增殖并诱导凋亡的实验研究[J]. 中国病理生理杂志, 2011, 27(6): 1084-1089.
[25]Fan S, Meng Q, Laterra JJ, et al. Ras effector pathways modulate scatter factor-stimulated NF-κB signaling and protection against DNA damage[J]. Oncogene, 2007, 26(33): 4774-4796.
[26]陈诗洪, 倪一虹, 庄向华, 等. 脂联素激活AKT抑制H2O2诱导的大鼠心肌细胞凋亡[J]. 中国病理生理杂志, 2010, 26(11): 2161-2164.
[27]Kashatus DF, Lim KH, Brady DC, et al. RALA and RALBP1 regulate mitochondrial fission at mitosis[J]. Nat Cell Biol, 2011, 13(9):1108-1115.
KnockdownofRALAbysiRNAinhibitscellproliferationandinducesapoptosisinhumanleukemicK562cells
LI Yu-min, ZHU Xue-jiao, GU Jing-yi, FEI Jia
(DepartmentofBiochemistryandMolecularBiology,SchoolofMedicine,JinanUniversity,Guangzhou510632,China.E-mail:efeijia@163.com)
AIM: To investigate the effect of siRNA-induced knockdown of v-ral simian leukemia viral oncogene homolog A(RALA) on proliferation and apoptosis of chronic myelogenous leukemia(CML) K562 cells.METHODSThe chemically synthesized siRNA targeting toRALAgene was transfected into K562 cells using LipofectamineTM2000. The proliferation and viability of K562 cells were detected by MTT assay and trypan blue dye exclusion. The expression levels of RALA mRNA and protein were determined by quantitative real-time PCR and Western blotting,respectively. The cell apoptosis was analyzed using flow cytometry by double staining with annexin V and propidium iodide, and the apoptotic morphological changes were detected by Hoechst 33258 staining.RESULTSRALA siRNA significantly down-regulatedRALAmRNA and protein expression in K562 cells(P<0.05). The proliferation of K562 cells inRALAsiRNA group was inhibited compared with control group(P<0.05). The apoptotic rate was much higher inRALAsiRNA group than that in negative control group(P<0.05). The apoptotic morphological changes were observed in the nuclei of K562 cells transfected with RALA siRNA.CONCLUSIONThe siRNA-mediated knockdown ofRALAresults in inhibition of proliferation and induction of apoptosis in K562 cells, indicating thatRALAmight be used as a potential therapeutic target in chronic myelogenous leukemia.
v-ral simian leukemia viral oncogene homolog A; siRNA; Leukemia; Cell proliferation; Apoptosis
R342.7
A
10.3969/j.issn.1000-4718.2012.04.009
猜你喜欢
军事文摘(2024年2期)2024-01-10 01:59:00
广东药科大学学报(2022年3期)2023-01-04 11:40:51
生物学通报(2022年1期)2022-11-22 08:12:18
南京林业大学学报(自然科学版)(2021年5期)2021-10-13 02:06:16
河南畜牧兽医(2017年12期)2017-11-13 04:05:18
健康管理(2016年2期)2016-05-30 21:36:03
广西林业科学(2016年3期)2016-03-16 05:43:25
中国科技信息(2015年6期)2015-11-10 03:35:44
中国医疗美容(2015年1期)2015-07-12 10:06:52
医学研究杂志(2015年9期)2015-07-01 17:27:46
