
过表达apoA-Ⅰ对BEL-7402细胞内脂质堆积的影响
2012-10-25 05:21王宇童
首都医科大学学报 2012年2期
覃 岭 王宇童
(首都医科大学基础医学院细胞生物学系,肝脏保护与再生调节北京市重点实验室,北京 100069)
肝脏是人体参与多种物质合成与代谢的重要器官,易受到如病毒、乙醇以及药物等多种毒物的损害。目前非酒精性脂肪性肝炎(non-alcoholic steatohepatitis,NASH)已成为全球肝病学家广为关注的一种肝病。NASH以脂肪变性与脂肪肝为特征,并且越来越多的研究[1-3]表明其与胰岛素耐受及代谢综合征相关。NASH的病因尚未明了,目前比较认同的观点是“二次打击”学说。有研究[4]结果显示,过多沉积于肝细胞的脂肪酸(free fatty acid,FFA)可能是诱发脂肪肝,造成NASH“第一次打击”的元凶之一,同时,FFA所引发的脂质过氧化反应,产生的超氧离子及自由基损伤是造成肝细胞“第二次打击”的重要原因。可见,FFA在NASH的发生发展过程中扮演着一个重要的角色。近期还有报道指出“第二次打击”还与自然杀伤 T 细胞(natural killer T,NKT)缺陷[5],T 细胞的调节异常[6]以及肠道微生物改变[7]有关。过量的脂质堆积在肝细胞是诱发肝炎发生的一个重要的因素[8],因此,有效地将过量的脂质转运出肝细胞是防治NASH的一个切入点。
ATP结合盒转运子A1(ATP-binding cassette transporter A1,ABCA1)可以将细胞内胆固醇和磷脂转运到细胞外的apoA-Ⅰ上,从而促进高密度脂蛋白(high density-lipoprotein,HDL)的形成。在此过程中,载脂蛋白 A-Ⅰ(apolipoprotein A-Ⅰ,apoA-Ⅰ)是接受胆固醇和磷脂的关键蛋白,此过程也是高密度脂蛋白合成的限速步骤[9-10]。特异性的蛋白酶、不饱和脂肪酸、胞内胆固醇的细胞毒作用可以降解ABCA1蛋白,近期还有报道[11]提出,不饱和脂肪酸通过抑制ABCA1的表达会导致脂质在肝癌细胞HepG2中堆积,apoA-Ⅰ可以与 ABCA1 结合[12-13],使 apoA-Ⅰ通过ABCA1上的PEST序列的磷酸化,抑制ABCA1钙蛋白酶降解,从而增加ABCA1蛋白质水平,促进脂质外流[14-16]。故本课题旨在研究:在过表达apoA-Ⅰ的细胞模型中,是否可以通过其对ABCA1的稳定作用来减少不饱和脂肪酸对ABCA1的降解来减少脂质在肝癌细胞中的堆积,从而探索一种新方法,通过apoA-Ⅰ/ABCA1途径改变细胞内脂质代谢来预防NASH的发生。
1 材料与方法
1.1 实验材料
实验室保存的BEL-7402细胞;Western blotting超敏发光液、游离脂肪酸超敏测定试剂盒、组织细胞总胆固醇酶法测定试剂盒、三酰甘油测定试剂盒、Hi-Gene转染试剂(北京普利莱基因技术有限公司);噻唑蓝(MTT)粉末(北京天来生物科技有限公司);glyceraldehyde-3-phosphate dehydrogenase(GAPDH)抗体(上海康辰生物有限公司);Plasmid Midi Kit(德国Qiagen 公司);pcDNA3.0 vector和 pcDNA3.0/apoA-Ⅰ(Invitrogen,Carlsbad,CA,美国);油红 O(上海斯红化工产品有限公司);蛋白酶抑制剂、棕榈酸(palmitic acid,PA)、油酸(oleic acid,OA)、牛血清白蛋白(bovine serum albumin,BSA)(Sigma公司,美国);apoA-Ⅰ抗体(Cell Signaling Technology公司,美国)。
1.2 实验方法
1)细胞培养及油红染色
在35 mm细胞培养皿中加入盖玻片,按4×105个细胞密度接种,用含10%的胎牛血清及1%的双抗,培养20~24 h,待细胞汇合率达到80%,分别用200 μmol/L 和400 μmol/L 的饱和脂肪酸 PA、不饱和脂肪酸 OA及 PA/OA混合物(比例为 PA∶OA=1∶2)[17]加入培养基中作用于细胞 6、12 及 24 h 后。依次将玻片取出,放入新的孔板中,用室温PBS冲洗3次,吸干多余水分,用10%中性甲醛固定细胞10 min,油红O工作液染色10 min,苏木精复染3 min,PBS冲洗10 min,50%甘油水溶液封片,显微镜下观察,细胞内的脂质为红色,细胞核为蓝色。
2)质粒的制备
按照Qiagene Plasmid Midi Kit说明书操作。首先挑取单克隆pcDNA3.0空载体以及pcDNA3.0/apoA-Ⅰ菌落,制备100 mL相应菌液,通过离心使细菌沉淀;其次,依次加入试剂盒中的P1、P2和P3裂解细菌,离心取上清;再通过试剂盒中的柱子使带负电的DNA黏附于柱子上,通过QBT以及QF将DNA洗涤、洗脱;最后用TE将pcDNA3.0空载体质粒以及pcDNA3.0/apoA-Ⅰ质粒溶解,测定DNA浓度。
按照HiGene转染试剂说明书的方法采用d=35 mm的培养皿转染真核细胞。①进行细胞悬液制备:用0.25%胰酶将细胞消化为单细胞悬液,按照4×105个细胞/皿准备细胞悬液备用;② 进行HiGene-DNA转染混合物制备:分别将3 μg质粒pcDNA3.0空载体(对照组)和质粒 pcDNA3.0/apoA-Ⅰ(实验组)稀释于200 μL无菌0.9%氯化钠注射液,振荡混匀后,取7.2 μL HiGene加到 DNA溶液中,立即振荡混匀,室温静置15 min后,全部加入到2 mL有血清无双抗的DMEM中备用;将备好的HiGene-DNA转染混合物加入到培养皿中后立即加入细胞悬液,实现铺板的同时对细胞进行转染。
4)测定细胞活力
将含有4×104个细胞的单细胞悬液100 μL分别接种于96孔板中,37℃、5%CO2培养细胞20~24 h后,分别更换含有200 μmol/L及400 μmol/L的不同脂肪酸的基础培养基,37℃、5%CO2培养细胞6、12及24 h。按照时间要求,每孔加入50 μL MTT液(2 mg/mL),37℃、5%CO2培养细胞继续培养3 h,吸出孔内培养液后,每孔加入150 μL DMSO液,避光,振荡10 min,使结晶物溶解。通过酶标仪,于490 nm波长处测各孔OD值。
5)蛋白定量及蛋白质印迹
在4℃条件下,加入裂解液提取人肝癌细胞系BEL-7402中的蛋白和细胞培养基中的蛋白。用BCA Protein Assay Kit测定样品蛋白含量。每个样本取50 μg蛋白进行SDS聚丙烯酰胺凝胶电泳并转至PVDF膜上,用含5% 脱脂奶粉的TBST溶液封闭4 h,加入1∶1 000 apoA-Ⅰ人单克隆抗体于4℃ 孵育过夜。TBST漂洗2次,每次5 min,加入1∶5 000辣根过氧化物酶标记的鼠抗人IgG,室温孵育1 h,TBST漂洗6次,每次5 min,GAPDH为内参,将膜用化学发光试剂孵育1 min,于X胶片上曝光,常规显影、定影。将结果用Genetools软件(Perkin Elmer)进行密度分析。
6)细胞内脂质的测定
分别按照三酰甘油酶法测定试剂盒、游离脂肪酸超敏测定试剂盒以及组织总胆固醇酶法测定试剂盒说明书中针对细胞测定的方法,采用d=35 mm细胞培养皿,分别按每106个细胞加0.1 mL相应裂解液的比例制备样品,按照说明书操作,得到相应的OD值,后依标准曲线,分别得到细胞内三酰甘油、游离脂肪酸以及总胆固醇的浓度,结果以mmol/L表示。
1.3 统计学方法
采用SPSS 13.0软件分析,组间比较采用单因素方差分析;以P<0.05为差异有统计学意义。
2 结果
2.1 油红O染色观察NASH细胞模型中脂质的形态
油红O染色结果表明,对照组BEL-7402细胞生长呈梭状或扁平多边形,胞质内无明显红染或淡染;模型组细胞中,尤其是用不饱和脂肪酸油酸(OA)及混合脂肪酸(PA:OA)作用的细胞质中广泛染色呈红色脂滴,大而多,而饱和脂肪酸棕榈酸(PA)与不饱和脂肪酸及混合脂肪酸相比,形成的脂质明显减少。另外,脂质形成随脂肪酸浓度以及作用时间的增加而增加(图1)。
记得2017年12月初,与儿子在绵阳。小子个子疯长,1米82了,我在他跟前,俨然是他的孩子了。吃饭,休息期间,儿子给我讲了很多他读过的书,有一些新科学,量子力学、暗物质之类,心生惭愧,也甚为骄傲。此外,儿子对文学和写作的理解,也使我自叹不如。更使我感动的是,在学校饭堂,儿子让我坐着,他给我打饭吃。那一刻,不由笑了,低头,老泪纵横。
2.2 MTT法检测不同脂肪酸对BEL-7402细胞活性的影响
采用200 μmol/L和400 μmol/L的不同脂肪酸处理BEL-7402细胞6、12和24 h后,MTT法测量细胞活性,结果显示:饱和脂肪酸棕榈酸的浓度越高,作用时间越长,对细胞活性的影响越大(图2)。
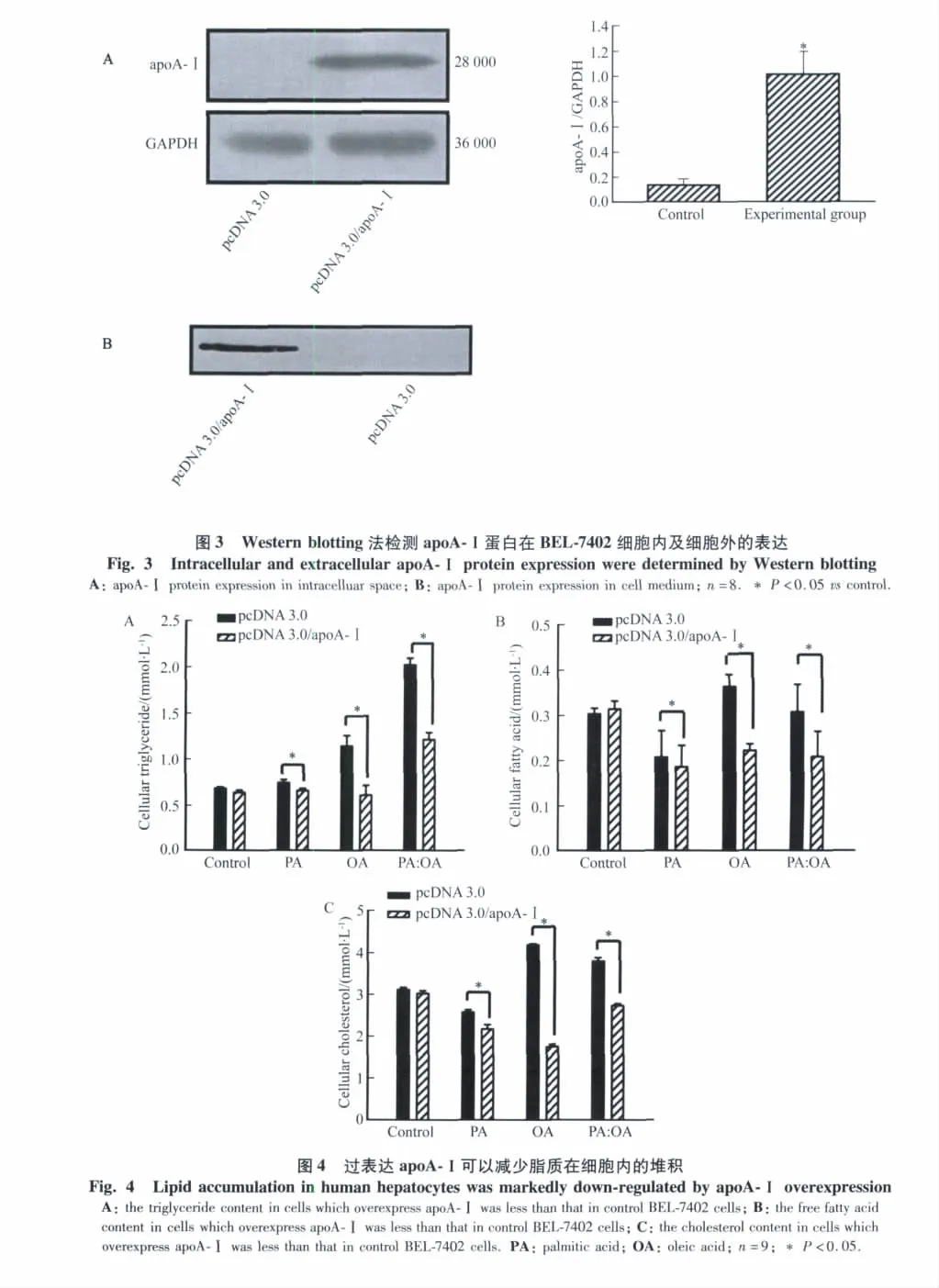
2.3 Western blotting方法检测apoA-Ⅰ的转染
转染了 pcDNA3.0/apoA-Ⅰ的细胞(实验组),通过与apoA-Ⅰ抗体作用,在28 000处出现明显的条带,而没有转染的pcDNA3.0空载体的细胞(对照组),则不见条带,说明apoA-Ⅰ转染成功(图3A)。转染了 apoA-Ⅰ的培养基中有apoA-Ⅰ存在,而转染空载体的细胞培养基中没有 apoA-Ⅰ(图 3B)。
2.4 apoA-Ⅰ对BEL-7402细胞内的脂质含量的影响
用200 μmol/L 浓度的 BSA、PA、OA、PA:OA 作用于正常细胞与转染了apoA-Ⅰ的细胞24 h,分别测定细胞内三酰甘油、游离脂肪酸以及胆固醇的含量。结果显示:与正常组(pcDNA3.0组)相比,过表达apoA-Ⅰ组(pcDNA3.0/apoA-Ⅰ组)细胞内各指标均减少。其中棕榈酸处理后的细胞脂质减少程度弱(图4),可能与棕榈酸本身在细胞内产生的脂质较少有关。

3 讨论
载脂蛋白A-Ⅰ是ABCA1的主要脂质接受体,外周组织细胞内过量的脂质可以通过ABCA1/apoA-Ⅰ途径,经胆固醇逆向转运促进细胞内胆固醇和磷脂的流出。该部分理论的应用主要用于防治动脉粥样硬化性心血管疾病,而在肝脏疾病领域应用的研究还比较少。本文主要探索ABCA1/apoA-Ⅰ途径在非酒精性脂肪性肝炎中的作用。
NAFLD是肝脏排除乙醇及病毒引起的肝脏从单纯脂肪变性发展到脂肪性肝炎、肝纤维化以及肝硬化的一系列病理综合征。但是其发病原因并不明确,目前被肝病学家广泛认同的机制是“二次打击”学说。近年研究[18-20]证实,游离脂肪酸(free-fatty acid,FFA)能激活使肝脏内氧化应激亢进的转录因子NFκB,这正是同NASH发病中的第一、二次打击均具相关性的重要因子。而它与肥胖、2型糖尿病以及高脂血症,尤其是高三酰甘油血症等代谢综合征有着密切的关系。FFA、三酰甘油以及胆固醇是脂质的重要成分,因此,研究如何预防NASH的发生,可以从减少肝细胞内脂质的堆积入手。众所周知,胆固醇和磷脂等脂质的转运过程主要在肝脏完成。在胆固醇逆向转运过程中,以apoA-Ⅰ为主的乏脂载脂蛋白可以将外周组织的胆固醇和磷脂转运到肝脏,经过肝脏进一步的代谢,最终以胆汁酸或胆汁的方式排出[21]。
据以往研究[22]显示,只有乏脂的apoA-Ⅰ结合于细胞膜上时,才能与ABCA1结合,并相互作用,转运胆固醇及磷脂,生成HDL。因此,在本实验中,过表达的apoA-Ⅰ只有释放到细胞外,才可以与ABCA1相互作用,将细胞内过量的脂质转运出细胞。这已在本实验中通过 Western blotting实验方法得到了验证:apoA-Ⅰ确实释放到细胞外。故该apoA-Ⅰ可以通过与细胞膜表面的ABCA1相互作用,行使其转运细胞内胆固醇及磷脂的功能。
本实验是通过用饱和、不饱和以及混合脂肪酸来构建含有脂质的BEL-7402细胞NASH模型,有相关研究[23]表明饱和脂肪酸棕榈酸可以诱导人肝癌HepG2细胞凋亡,本实验中使用的是人肝癌 BEL-7402细胞,棕榈酸是否也会导致该细胞株的凋亡,以及什么样的剂量以及作用时间导致其凋亡我们还不清楚,故本实验分别用200 μmol/L和400 μmol/L的不同脂肪酸处理BEL-7402细胞6、12和24 h后,用MTT法测量细胞活性。原则上选择最小剂量以及最短作用时间作为构建NASH细胞模型的实验条件,因此本实验选择200 μmol/L浓度为最佳浓度,24 h为最佳作用时间。另外还有报道[24]指出,由不饱和脂肪酸衍生出来的含有双键的二酰甘油可进一步激活蛋白激酶Cδ(PKCδ),使ABCA1中丝氨酸的磷酸化水平增加,从而降低ABCA1蛋白稳定性,使蛋白降解速度加快。我们在前期研究[11]中发现,在人的肝癌细胞系HepG2细胞中,不饱和脂肪酸通过加快ABCA1的降解来抑制ABCA1蛋白质水平,ABCA1的表达直接影响到细胞内的FFA和三酰甘油的形成,同时在NASH大鼠肝脏中也看到ABCA1蛋白质水平的下降。而本文结果显示,在BEL-7402细胞中,过表达apoA-Ⅰ后,由不饱和脂肪酸引起的细胞内脂质蓄积程度比对照组细胞明显减轻。这与杨峻浩等[25]用apoA-Ⅰ处理单核巨噬细胞(human acute monocytic leukemia cell line,THP-1)源性泡沫细胞得到的结果一致。这些结果表明,apoA-Ⅰ与ABCA1相互作用一方面可以增强细胞ABCA1的脂质转运功能,另一方面apoA-Ⅰ还可以通过和ABCA1结合,对ABCA1蛋白质起到稳定作用,从而提高细胞膜上ABCA1的蛋白质水平[26]。近期有实验结果[27]显示,apoA-Ⅰ可降低THP-1巨噬细胞源性泡沫细胞内胆固醇聚积的机制可能是通过PKA信号途径使细胞ABCA1蛋白质水平增加,而在本实验中,apoA-Ⅰ在肝癌细胞内降低三酰甘油、游离脂肪酸以及胆固醇的聚积可能也是通过该途径实现的。脂肪酸合成酶、乙酰辅酶A羧化酶等与脂质合成密切相关,有文献[28]报道,氧甾酮作为LXR的配体,可调节这些重要酶的转录水平。因此我们还可以通过过量表达apoA-Ⅰ,从转录水平上,这些下调与脂质合成有关的酶的表达,从而降低细胞内脂质的堆积。更好地了解ABCA1/apoA-Ⅰ介导脂质转运途径及其调节机制,通过调控该途径相关基因来防治NASH这一人类面临的新的挑战。
[1]Matteoni C A,Younossi Z M,Gramlich T,et al.Nonalcoholic fatty liver disease:a spectrum of clinical and pathological severity[J].Gastroenterology,1999,116(6):1413-1419.
[2]Okamoto M,Takeda Y,Yoda Y,et al.The association of fatty liver and diabetes risk[J].J Epidemiol,2003,13(1):15-21.
[3]Hamaguchi M,Kojima T,Takeda N,et al.The metabolic syndrome as a predictor of nonalcoholic fatty liver disease[J].Ann Intern Med,2005,143(10):722-728.
[4]Seki S,Kitada T,Yamada T,et al.In situ detection of lipid peroxidation and oxidative DNA damage in non-alcoholic fatty liver diseases[J].J Hepatol,2002,37(1):56-62.
[5]Kremer M,Thomas E,Milton R J,et al.Kupffer cell and interleukin-12-dependent loss of natural killer T cells in hepatosteatosis[J].Hepatology,2010,51(1):130-141.
[6]Ma X,Hua J,Mohamood A R,et al.A high-fat diet and regulatory T cells influence susceptibility to endotoxin-induced liver injury[J].Hepatology,2007,46(5):1519-1529.
[7]Musso G,Gambino R,Cassader M.Gut microbiota as a regulator of energy homeostasis and ectopic fat deposition:mechanisms and implications for metabolic disorders[J].Curr Opin Lipidol,2010,21(1):76-83.
[8]Sheth S G,Gordon F D,Chopra S.Non alcoholic steatohepatitis[J].Ann Intern Med,1997,126(2):137-145.
[9]Joyce C,Freeman L,Brewer H B Jr,et al.Study of ABCA1 function in transgenic mice[J].Arterioscler Thromb Vasc Biol,2003,23(6):965-971.
[10]Aiello R J,Brees D,Francone O L.ABCA1-deficient mice:insights into the role of monocyte lipid efflux in HDL formation and inflammation[J].Arterioscler Thromb Vasc Biol,2003,23(6):972-980.
[11]Yang Y,Jiang Y,Wang Y,et al.Suppression of ABCA1 by unstaurated fatty acids leads to lipid accumulation in HepG2 cells[J].Biochimie,2010,92(8):958-963.
[12]Rubin E M,Spangler E.A,Verstuyft J G,et al.Inhibition of early atherogenesis in transgenic mice by human apolipoprotein AI[J].Nature,1991,353(6341):265-267.
[13]Cole T G,Nowatzke W L,Bisgaier C L,et al.Method-dependent changes in ”HDL-cholesterol”with recombinant apolipoprotein A-Ⅰ(Milano)infusion in healthy volunteers[J].Clin Chem,2002,48(4):680-681.
[14]Wang N,Chen W,Linsel-Nitschke P,et al.A PEST sequence in ABCA1 regulates degradation by calpain protease and stabilization of ABCA1 by apoA-Ⅰ[J].J Clin Invest,2003,111(1):99-107.
[15]Tang C K,Tang G H,Yi G H,et al.Effect of apolipoprotein A-Ⅰon ATP binding cassette transporter A1 degradation and cholesterol efflux in THP-1 macrophage-derived foam cells[J].Acta Biochim Biophys Sin(Shanghai),2004,36(3):218-226.
[16]Duong P T,Collins H L,Nickel M,et al.Characterization of nascent HDL particles and microparticles formed by ABCA1-mediated efflux of cellular lipids to apoA-Ⅰ[J].J Lipid Res,2006,47(4):832-843.
[17]Liu J,Li Y M,Chen S H,et al.An in vitro hepatic steatosis cell model for study of non-alcoholic fatty liver disease[J].Zhejiang Da Xue Xue Bao Yi Xue Ban,2009,38(6):626-629.
[18]Zhang L,Ji J,Zhu X Y,et al.Palmitic acid induces apoptosis in human hepatoma cell line,HepG2 cells[J].Zhongguo Yi Xue Ke Xue Yuan Xue Bao,2004,26(6):671-676.
[19]Saleh J,Sniderman A D,Cianflone K.Regulation of plasma fatty acid metabolism[J].Clin Chim Acta,1999,286(1-2):163-180.
[20]Mehta K,Van Thiel D H,Shah N,et al.Nonalcoholic fatty liver disease:pathogenesis and the role of antioxidants[J].Nutr Rev,2002,60(9):289-293.
[21]Oram J F,Yokoyama S.Apolipoprotein-mediated removal of cellular cholesterol and phospholipids[J].J Lipid Res,1996,37(12):2473-2491.
[22]Fitzgerald M L,Morris A L,Chroni A,et al.ABCA1 and amphipathic apolipoproteins form high-affinity molecular complexes required for cholesterol efflux[J].J Lipid Res,2004,45(2):287-294.
[23]Matteoni C A,Younossi Z M,Gramlich T,et al.Nonalcoholic fatty liver disease:a spectrum of clinical and pathological severity[J].Gastroenterology,1999,116(6):1413-1419.
[24]Wang Y,Oram J F.Unsaturated fatty acids inhibit cholesterol efflux from macrophages by increasing degradation of ATP-binding cassette transporter A1[J].J Biol Chem,2002,277(7):5692-5697.
[25]杨峻浩,代小艳,欧翔,等.载脂蛋白A-Ⅰ通过PKA信号途径影响ABCA1的表达与功能[J].生物化学与生物物理进展,2007,34(6):611-619.
[26]Wang N,Chen W,Linsel-Nitschke P,et al.A PEST sequence in ABCA1 regulates degradation by calpain protease and stabilization of ABCA1 by apoA-Ⅰ[J].J Clin Invest,2003,111(1):99-107.
[27]Liu X H,Xiao J,Mo Z C,et al.Contribution of D4-F to ABCA1 expression and cholesterol efflux in THP-1 macrophage-derived foam cells[J].J Cardiovasc Pharmacol,2010,56(3):309-319.
[28]Choe S S,Choi A H,Lee J W,et al.Chronic activation of liver X receptor induces beta-cell apoptosis through hyperactivation of lipogenesis:liver X receptor-mediated lipotoxicity in pancreatic beta-cells[J].Diabetes,2007,56(6):1534-1543.
猜你喜欢
中国医药科学(2021年18期)2021-01-03
天然产物研究与开发(2018年9期)2018-10-08
能源(2017年7期)2018-01-19
中学科技(2017年11期)2017-12-26
中学科技(2016年5期)2016-05-12
中国民族医药杂志(2016年4期)2016-05-09
武警医学(2016年6期)2016-03-13
合成化学(2015年10期)2016-01-17
西安交通大学学报(医学版)(2015年2期)2015-02-28
化工生产与技术(2014年4期)2014-02-27
